

Crystal packing modifies ligand binding affinity: the case of aldose reductase
- PMID: 22752989
- PMCID: PMC4671318
- DOI: 10.1002/prot.24136
Crystal packing modifies ligand binding affinity: the case of aldose reductase
Abstract
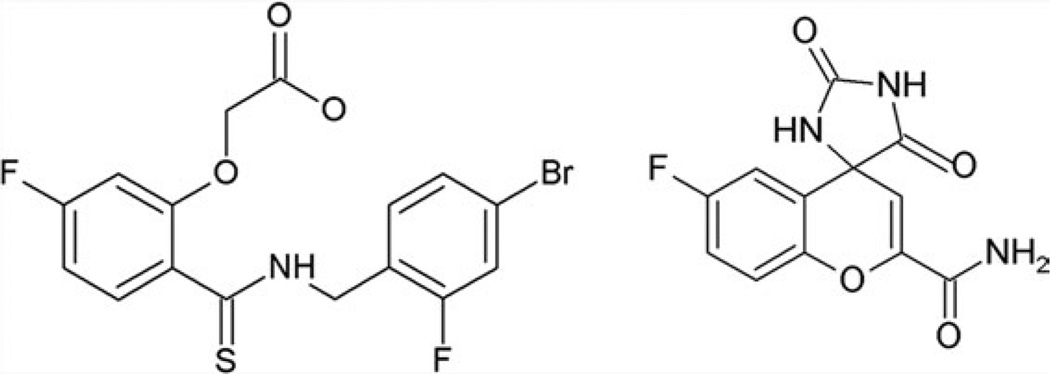
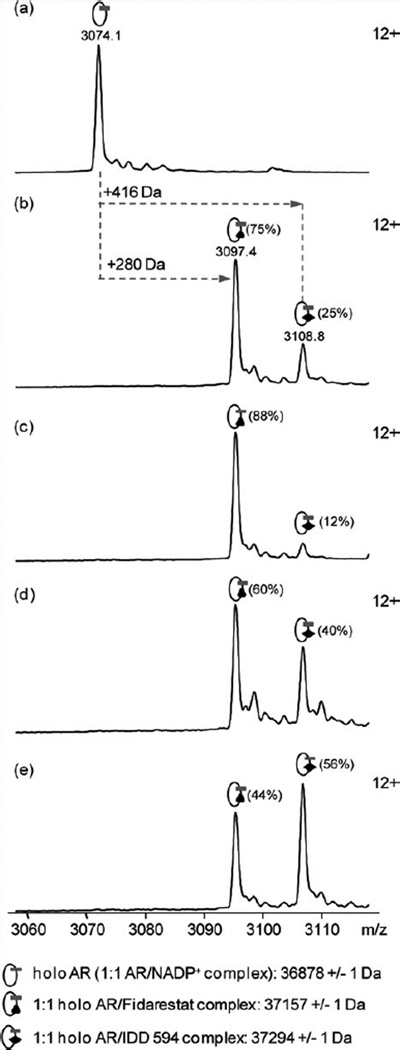
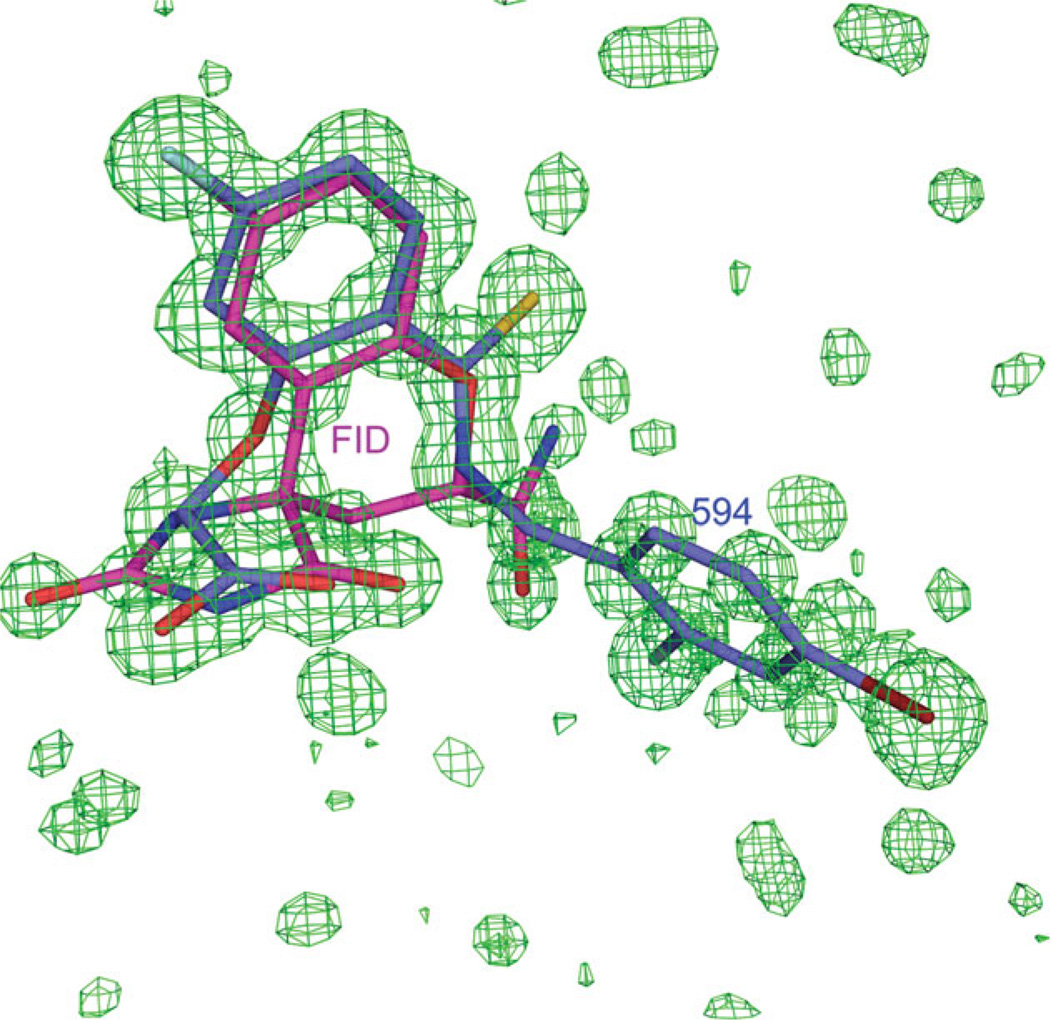
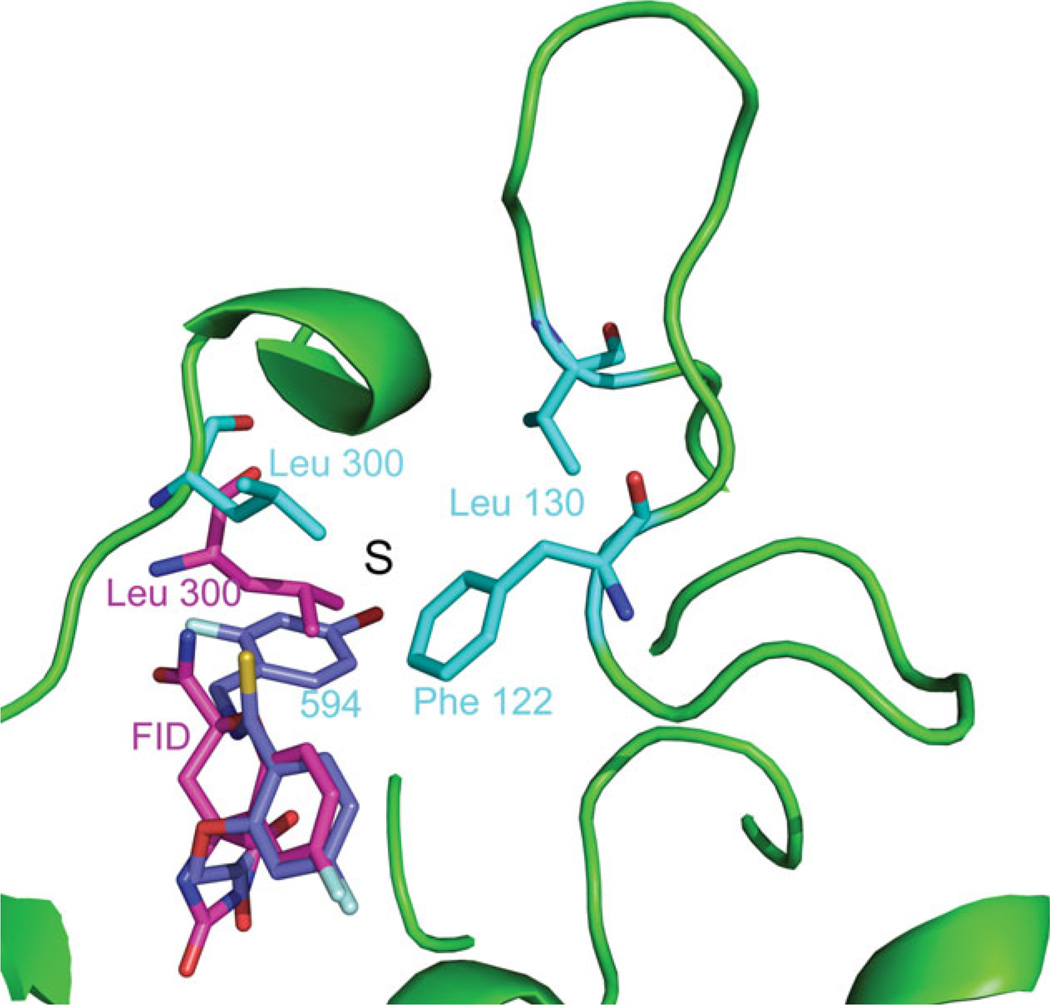
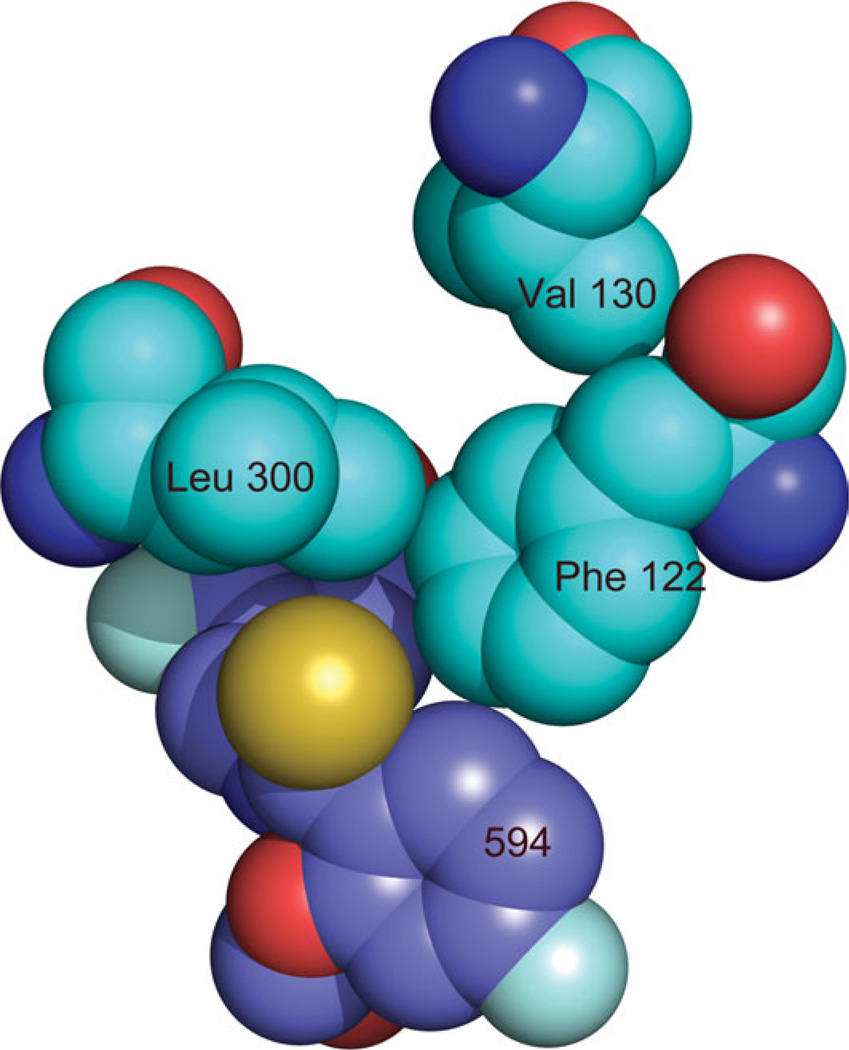
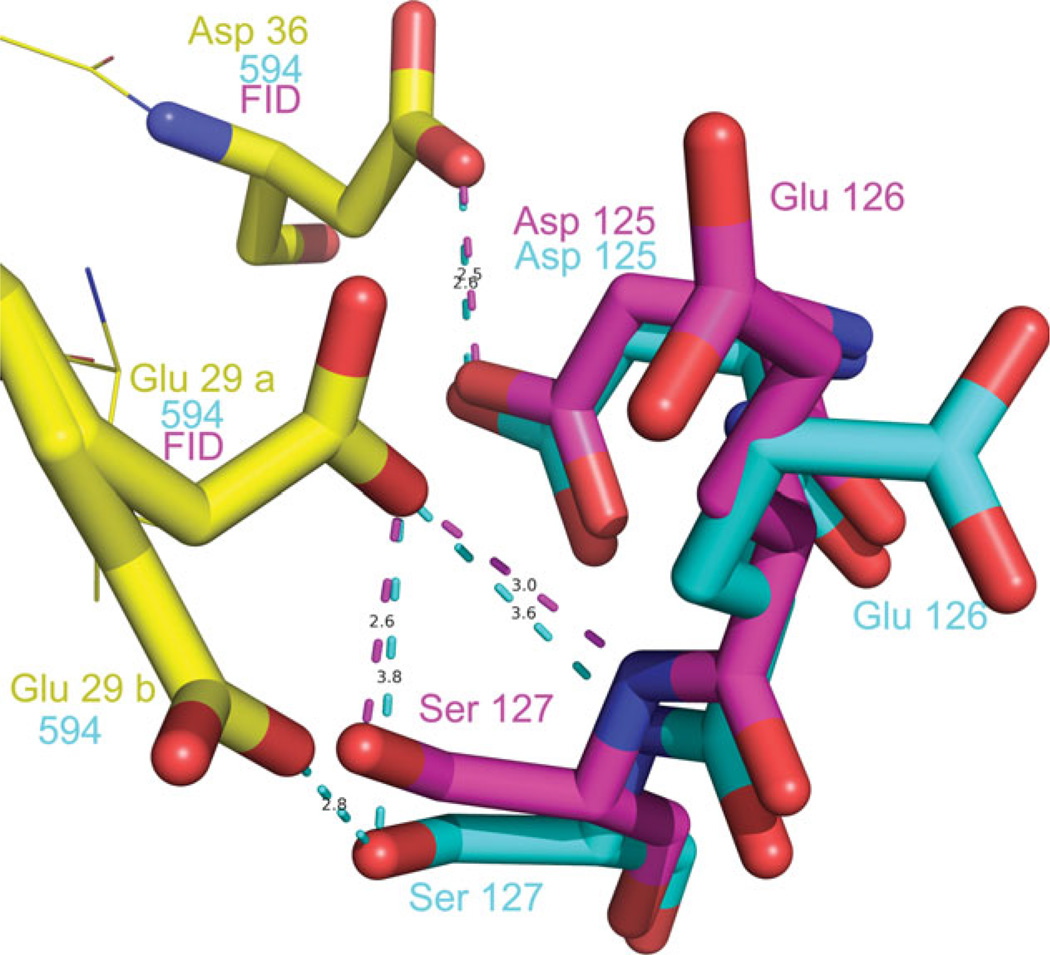
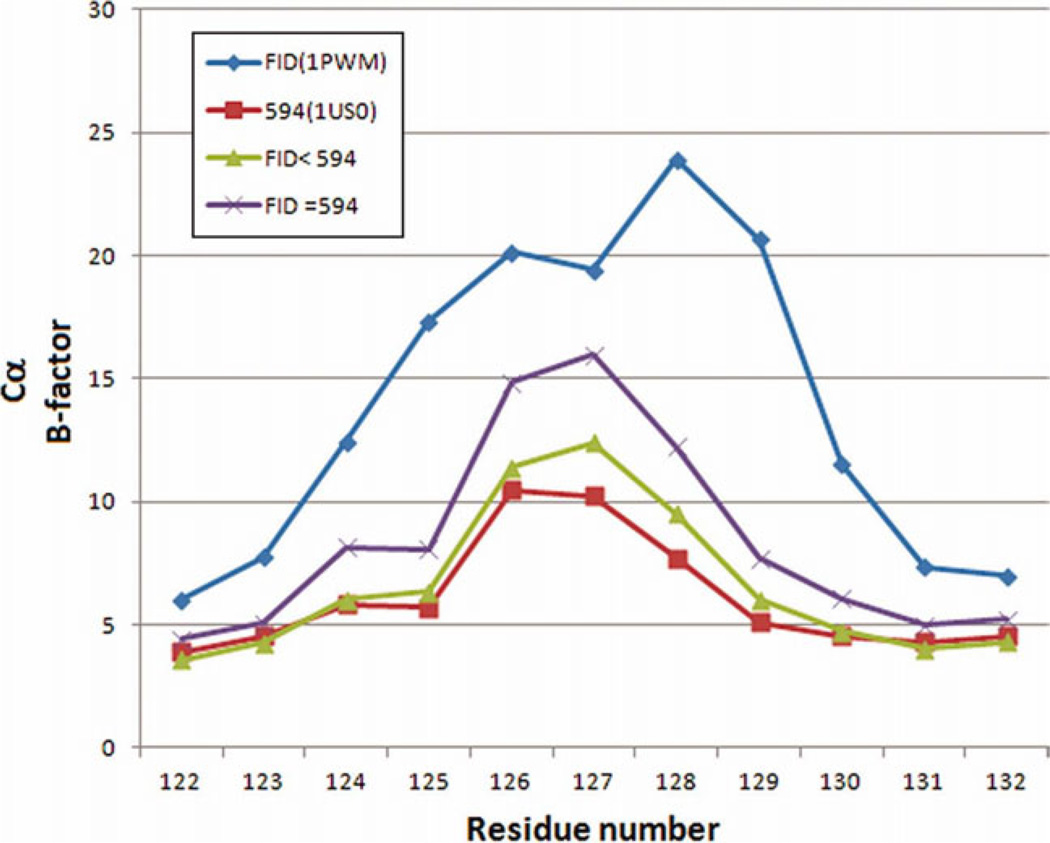
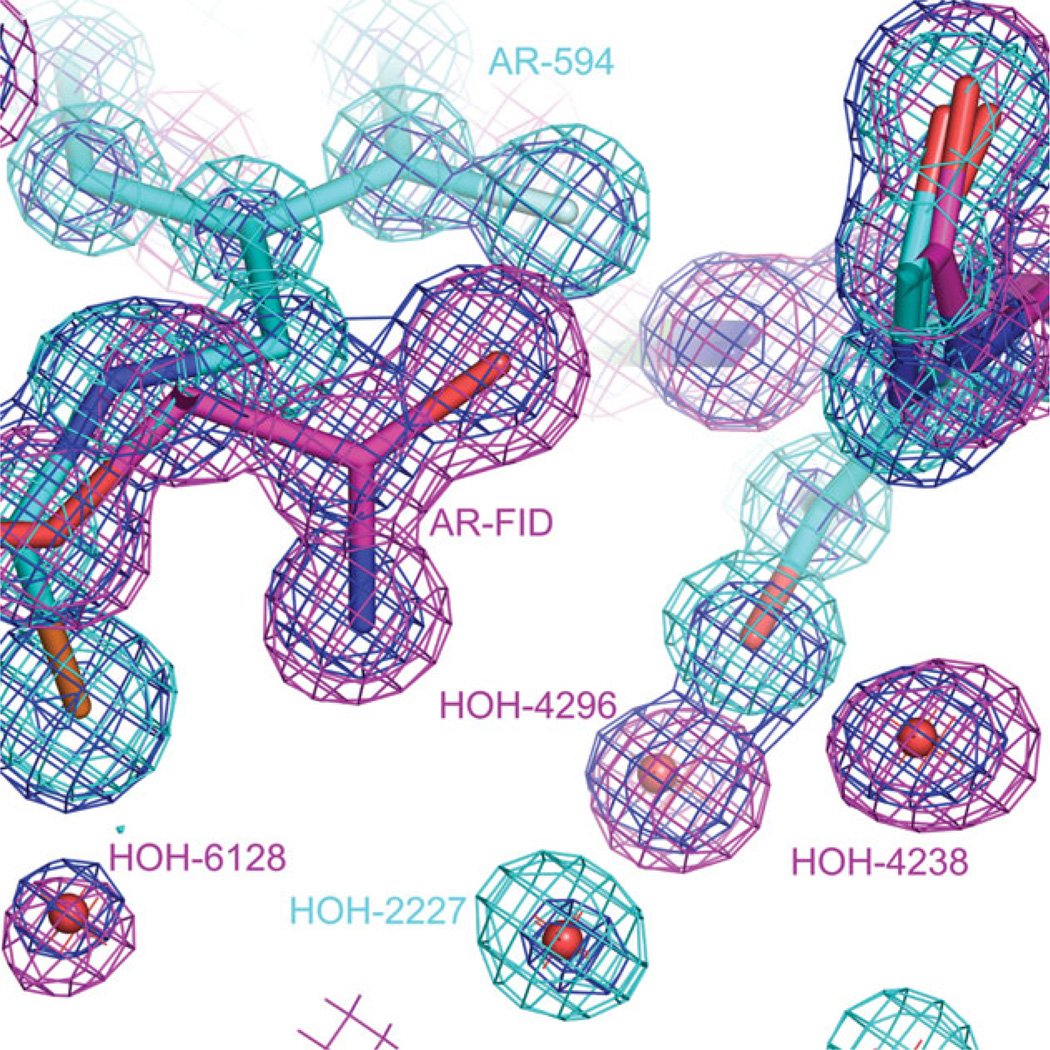
The relationship between the structures of protein-ligand complexes existing in the crystal and in solution, essential in the case of fragment-based screening by X-ray crystallography (FBS-X), has been often an object of controversy. To address this question, simultaneous co-crystallization and soaking of two inhibitors with different ratios, Fidarestat (FID; K(d) = 6.5 nM) and IDD594 (594; K(d) = 61 nM), which bind to h-aldose reductase (AR), have been performed. The subatomic resolution of the crystal structures allows the differentiation of both inhibitors, even when the structures are almost superposed. We have determined the occupation ratio in solution by mass spectrometry (MS) Occ(FID)/Occ(594) = 2.7 and by X-ray crystallography Occ(FID)/Occ(594) = 0.6. The occupancies in the crystal and in solution differ 4.6 times, implying that ligand binding potency is influenced by crystal contacts. A structural analysis shows that the Loop A (residues 122-130), which is exposed to the solvent, is flexible in solution, and is involved in packing contacts within the crystal. Furthermore, inhibitor 594 contacts the base of Loop A, stabilizing it, while inhibitor FID does not. This is shown by the difference in B-factors of the Loop A between the AR-594 and AR-FID complexes. A stable loop diminishes the entropic energy barrier to binding, favoring 594 versus FID. Therefore, the effect of the crystal environment should be taken into consideration in the X-ray diffraction analysis of ligand binding to proteins. This conclusion highlights the need for additional methodologies in the case of FBS-X to validate this powerful screening technique, which is widely used.
Copyright © 2012 Wiley Periodicals, Inc.
Figures
References
-
- Danley DE. Crystallization to obtain protein-ligand complexes for structure-aided drug design. Acta Crystallogr D Biol Crystallogr. 2006;62(Part 6):569–575. - PubMed
-
- Rondeau J, Klebe G, Podjarny A. Ligand binding: the crystallographic approach. In: Podjarny A, Dejaegere A, Kieffer B, editors. Biophysical approaches determining ligand binding to biomolecular targets: detection, measurement and modelling. modelling. Vol. 1: Biomolecular sciences. London: RSC Publishing; 2011. pp. 56–135.
-
- Blundell TL, Jhoti H, Abell C. High-throughput crystallography for lead discovery in drug design. Nat Rev Drug Discov. 2002;1:45–54. - PubMed
-
- Skarzynski T, Thorpe J. Industrial perspective on X-ray data collection and analysis. Acta Crystallogr D Biol Crystallogr. 2006;62(Part 1):102–107. - PubMed
-
- Cimmperman P, Matulis D. Protein thermal denaturation measurements via a fluorescent dye. In: Podjarny A, Dejaegere A, Kieffer B, editors. Biophysical approaches determining ligand binding to biomolecular targets: detection, measurement and modelling. Vol. 1: Biomolecular sciences. London: RSC Publishing; 2011. pp. 247–274.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
