

In silico method for modelling metabolism and gene product expression at genome scale
- PMID: 22760628
- PMCID: PMC3827721
- DOI: 10.1038/ncomms1928
In silico method for modelling metabolism and gene product expression at genome scale
Abstract
Transcription and translation use raw materials and energy generated metabolically to create the macromolecular machinery responsible for all cellular functions, including metabolism. A biochemically accurate model of molecular biology and metabolism will facilitate comprehensive and quantitative computations of an organism's molecular constitution as a function of genetic and environmental parameters. Here we formulate a model of metabolism and macromolecular expression. Prototyping it using the simple microorganism Thermotoga maritima, we show our model accurately simulates variations in cellular composition and gene expression. Moreover, through in silico comparative transcriptomics, the model allows the discovery of new regulons and improving the genome and transcription unit annotations. Our method presents a framework for investigating molecular biology and cellular physiology in silico and may allow quantitative interpretation of multi-omics data sets in the context of an integrated biochemical description of an organism.
Figures



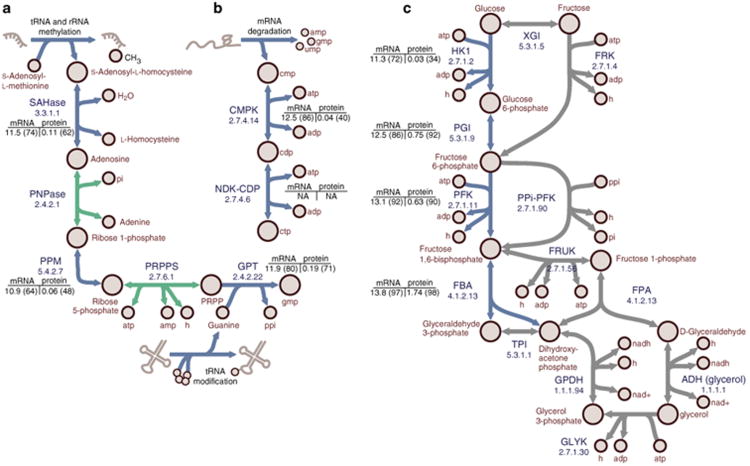
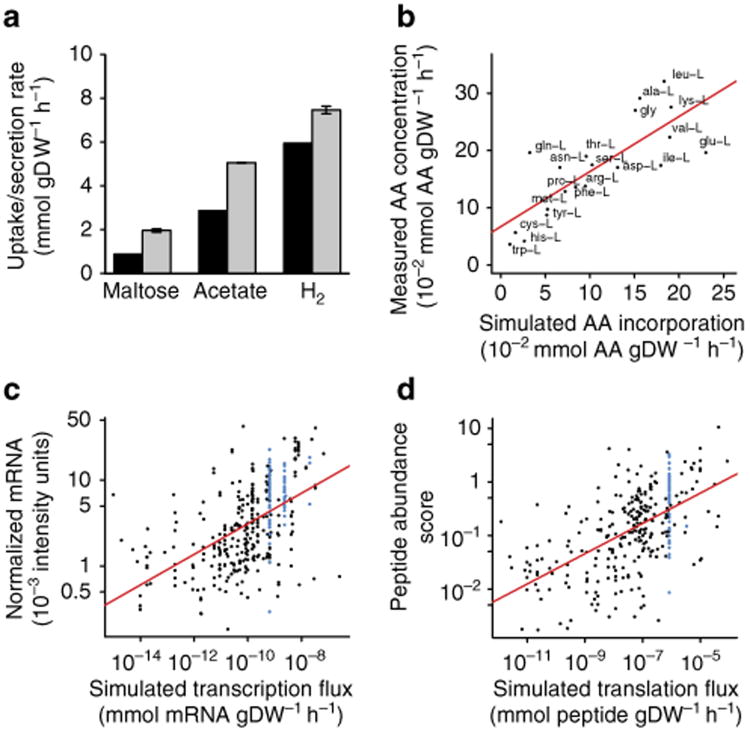
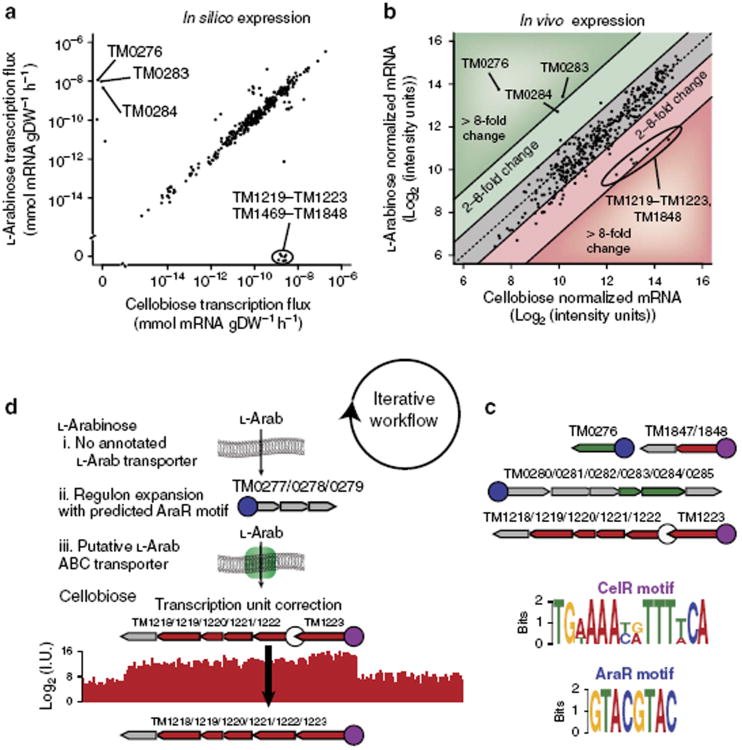
References
-
- Otero JM, Nielsen J. Industrial systems biology. Biotechnol Bioeng. 2010;105:439–460. - PubMed
-
- Palsson B, Zengler K. The challenges of integrating multi-omic data sets. Nat Chem Biol. 2010;6:787–789. - PubMed
-
- Mahadevan R, Palsson BO, Lovley DR. In situ to in silico and back: elucidating the physiology and ecology of Geobacter spp. using genome-scale modelling. Nat Rev Microbiol. 2011;9:39–50. - PubMed
Publication types
MeSH terms
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
