

Aldehyde dehydrogenase type 2 activation by adenosine and histamine inhibits ischemic norepinephrine release in cardiac sympathetic neurons: mediation by protein kinase Cε
- PMID: 22761303
- PMCID: PMC3464041
- DOI: 10.1124/jpet.112.196626
Aldehyde dehydrogenase type 2 activation by adenosine and histamine inhibits ischemic norepinephrine release in cardiac sympathetic neurons: mediation by protein kinase Cε
Abstract
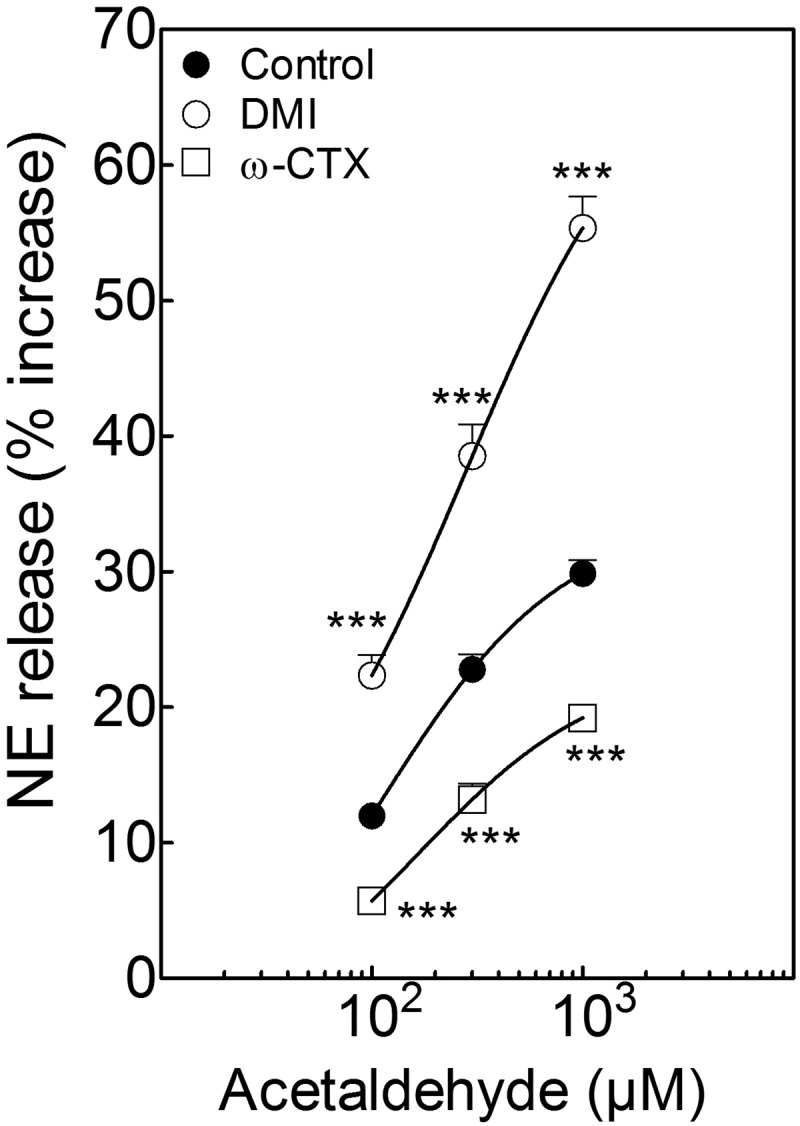
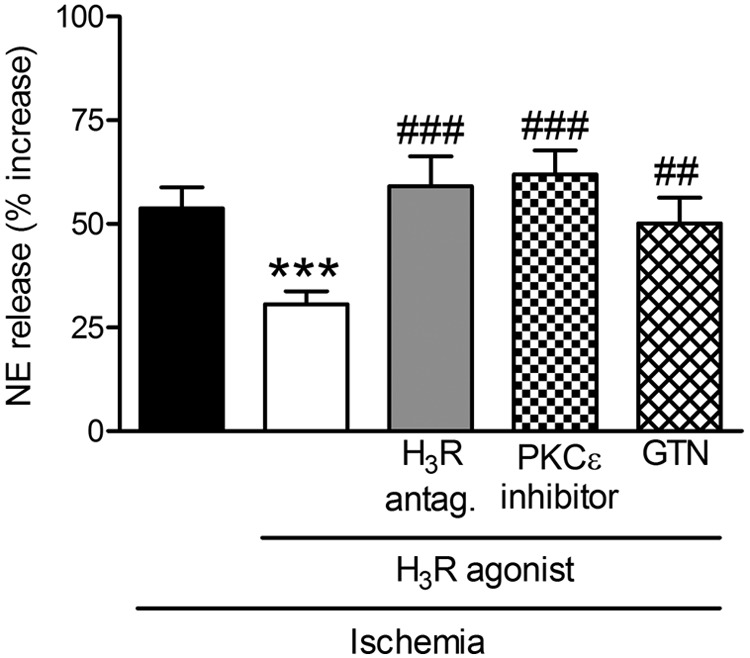
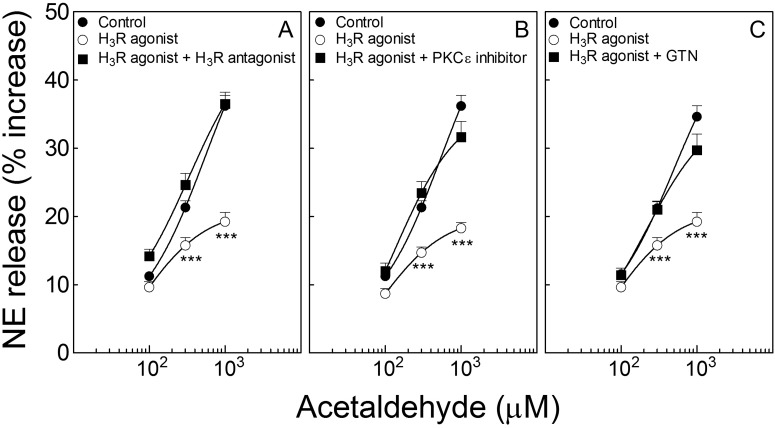
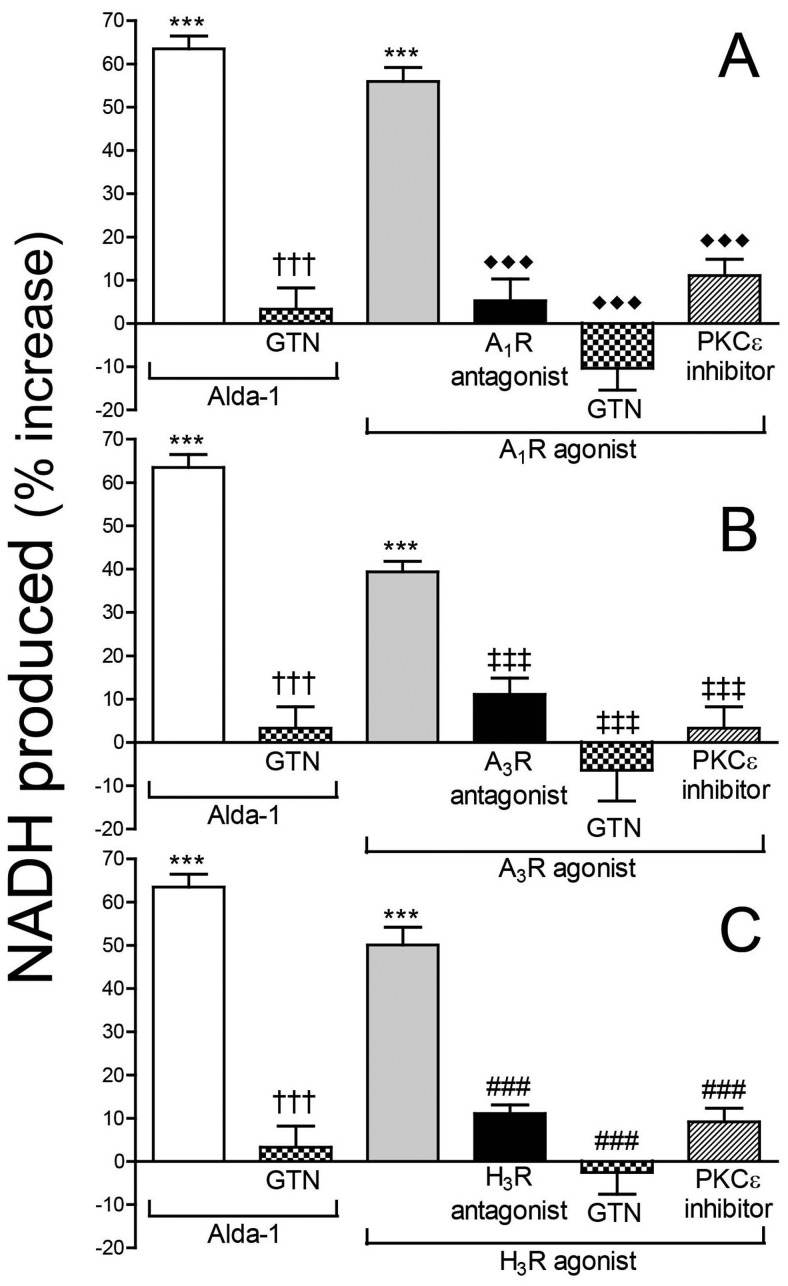
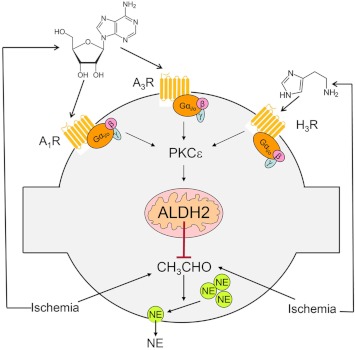
During myocardial ischemia/reperfusion, lipid peroxidation leads to the formation of toxic aldehydes that contribute to ischemic dysfunction. Mitochondrial aldehyde dehydrogenase type 2 (ALDH2) alleviates ischemic heart damage and reperfusion arrhythmias via aldehyde detoxification. Because excessive norepinephrine release in the heart is a pivotal arrhythmogenic mechanism, we hypothesized that neuronal ALDH2 activation might diminish ischemic norepinephrine release. Incubation of cardiac sympathetic nerve endings with acetaldehyde, at concentrations achieved in myocardial ischemia, caused a concentration-dependent increase in norepinephrine release. A major increase in norepinephrine release also occurred when sympathetic nerve endings were incubated in hypoxic conditions. ALDH2 activation substantially reduced acetaldehyde- and hypoxia-induced norepinephrine release, an action prevented by inhibition of ALDH2 or protein kinase Cε (PKCε). Selective activation of G(i/o)-coupled adenosine A(1), A(3), or histamine H(3) receptors markedly inhibited both acetaldehyde- and hypoxia-induced norepinephrine release. These effects were also abolished by PKCε and/or ALDH2 inhibition. Moreover, A(1)-, A(3)-, or H(3)-receptor activation increased ALDH2 activity in a sympathetic neuron model (differentiated PC12 cells stably transfected with H(3) receptors). This action was prevented by the inhibition of PKCε and ALDH2. Our findings suggest the existence in sympathetic neurons of a protective pathway initiated by A(1)-, A(3)-, and H(3)-receptor activation by adenosine and histamine released in close proximity of these terminals. This pathway comprises the sequential activation of PKCε and ALDH2, culminating in aldehyde detoxification and inhibition of hypoxic norepinephrine release. Thus, pharmacological activation of PKCε and ALDH2 in cardiac sympathetic nerves may have significant protective effects by alleviating norepinephrine-induced life-threatening arrhythmias that characterize myocardial ischemia/reperfusion.
Figures
Similar articles
-
Histamine H4-receptors inhibit mast cell renin release in ischemia/reperfusion via protein kinase C ε-dependent aldehyde dehydrogenase type-2 activation.J Pharmacol Exp Ther. 2014 Jun;349(3):508-17. doi: 10.1124/jpet.114.214122. Epub 2014 Apr 2. J Pharmacol Exp Ther. 2014. PMID: 24696042 Free PMC article.
-
Aldehyde dehydrogenase activation prevents reperfusion arrhythmias by inhibiting local renin release from cardiac mast cells.Circulation. 2010 Aug 24;122(8):771-81. doi: 10.1161/CIRCULATIONAHA.110.952481. Epub 2010 Aug 9. Circulation. 2010. PMID: 20697027 Free PMC article.
-
Salvaging the Ischemic Heart: Gi-Coupled Receptors in Mast Cells Activate a PKCε/ALDH2 Pathway Providing Anti-RAS Cardioprotection.Curr Med Chem. 2018;25(34):4416-4431. doi: 10.2174/0929867325666180214115127. Curr Med Chem. 2018. PMID: 29446730 Review.
-
Natriuretic peptide-induced catecholamine release from cardiac sympathetic neurons: inhibition by histamine H3 and H4 receptor activation.J Pharmacol Exp Ther. 2012 Dec;343(3):568-77. doi: 10.1124/jpet.112.198747. Epub 2012 Aug 24. J Pharmacol Exp Ther. 2012. PMID: 22923736 Free PMC article.
-
Aldehyde dehydrogenase 2 in cardiac protection: a new therapeutic target?Trends Cardiovasc Med. 2009 Jul;19(5):158-64. doi: 10.1016/j.tcm.2009.09.003. Trends Cardiovasc Med. 2009. PMID: 20005475 Free PMC article. Review.
Cited by
-
Modulation of Ion Channels in the Superior Cervical Ganglion Neurons by Myocardial Ischemia and Fluvastatin Treatment.Front Physiol. 2018 Sep 10;9:1157. doi: 10.3389/fphys.2018.01157. eCollection 2018. Front Physiol. 2018. PMID: 30246810 Free PMC article.
-
Targeting aldehyde dehydrogenase 2: new therapeutic opportunities.Physiol Rev. 2014 Jan;94(1):1-34. doi: 10.1152/physrev.00017.2013. Physiol Rev. 2014. PMID: 24382882 Free PMC article. Review.
-
Selective cytoprotective effect of histamine on doxorubicin-induced hepatic and cardiac toxicity in animal models.Cell Death Discov. 2015 Dec 21;1:15059. doi: 10.1038/cddiscovery.2015.59. eCollection 2015. Cell Death Discov. 2015. PMID: 27551485 Free PMC article.
-
Increased expression of microRNA-378a-5p in acute ethanol exposure of rat cardiomyocytes.Cell Stress Chaperones. 2017 Mar;22(2):245-252. doi: 10.1007/s12192-016-0760-y. Epub 2017 Feb 3. Cell Stress Chaperones. 2017. PMID: 28160209 Free PMC article.
References
-
- Aloyo VJ, McIlvain HB, Bhavsar VH, Roberts J. (1991) Characterization of norepinephrine accumulation by a crude synaptosomal-mitochondrial fraction isolated from rat heart. Life Sci 48:1317–1324 - PubMed
-
- Carini R, De Cesaris MG, Spendore R, Albano E. (2000) Ethanol potentiates hypoxic liver injury: role of hepatocyte Na+ overload. Biochim Biophys Acta 1502:508–514 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
