

Antagonism of miR-21 reverses epithelial-mesenchymal transition and cancer stem cell phenotype through AKT/ERK1/2 inactivation by targeting PTEN
- PMID: 22761812
- PMCID: PMC3382593
- DOI: 10.1371/journal.pone.0039520
Antagonism of miR-21 reverses epithelial-mesenchymal transition and cancer stem cell phenotype through AKT/ERK1/2 inactivation by targeting PTEN
Abstract
Background: Accumulating evidence suggested that epithelial-mesenchymal transition (EMT) and cancer stem cell (CSC) characteristics, both of which contribute to tumor invasion and metastasis, are interrelated with miR-21. MiR-21 is one of the important microRNAs associated with tumor progression and metastasis, but the molecular mechanisms underlying EMT and CSC phenotype during miR-21 contributes to migration and invasion of breast cancer cells remain to be elucidated.
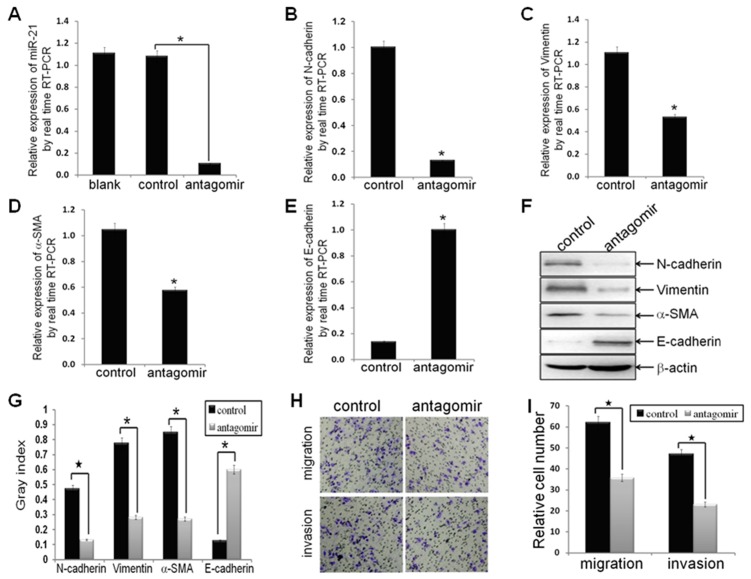
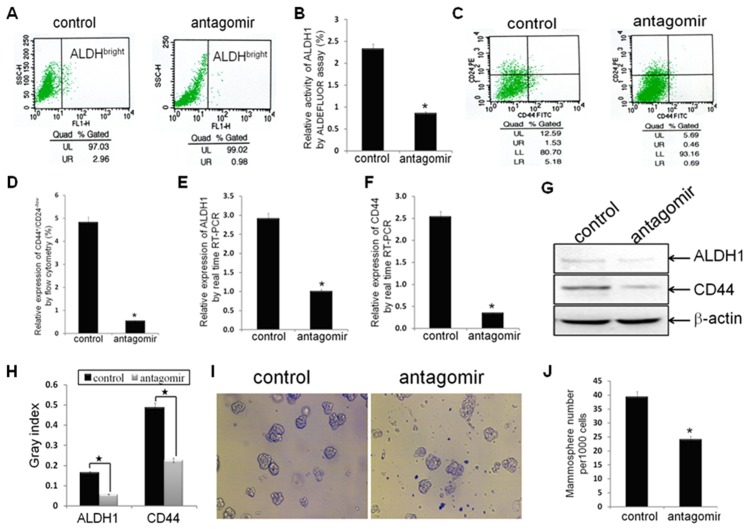
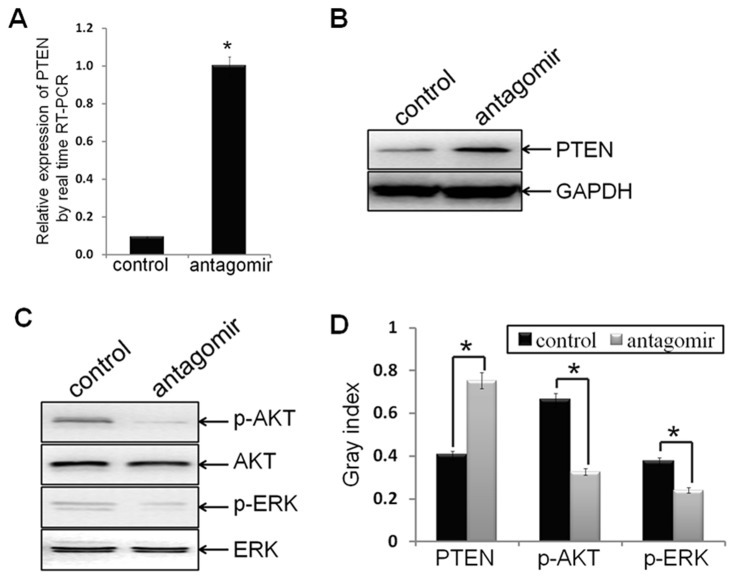
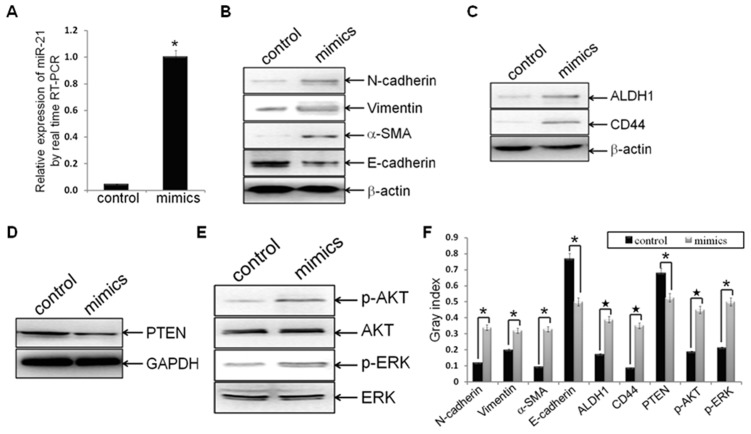
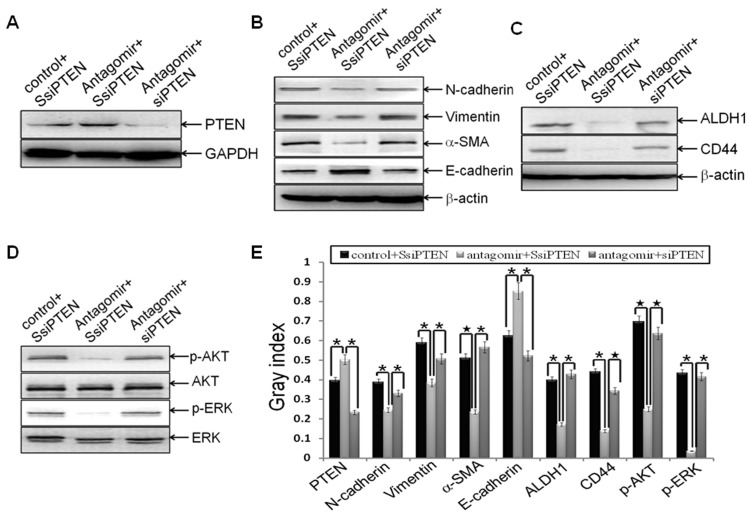
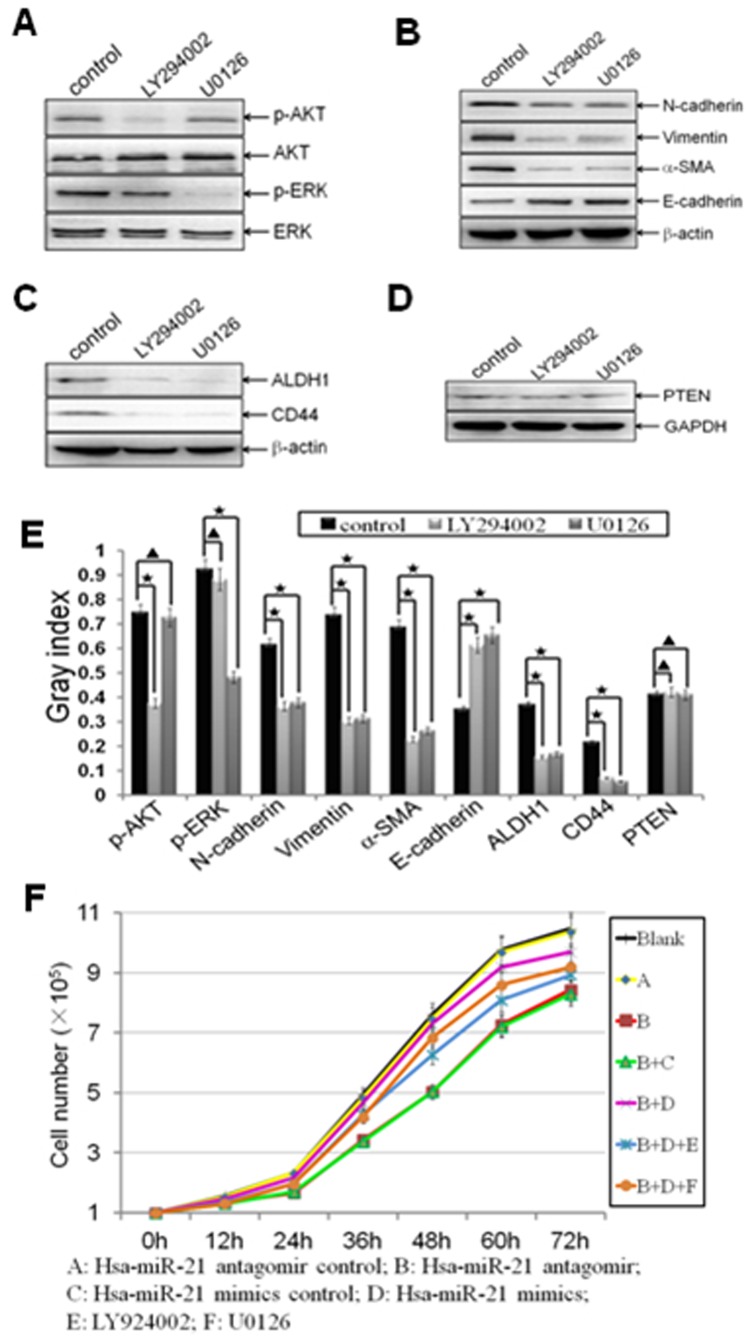
Methodology/principal findings: In this study, MDA-MB-231/anti-miR-21 cells were established by transfected hsa-miR-21 antagomir into breast cancer MDA-MB-231 cells. EMT was evaluated by the changes of mesenchymal cell markers (N-cadherin, Vimentin, and alpha-SMA), epithelial cell marker (E-cadherin), as well as capacities of cell migration and invasion; CSC phenotype was measured using the changes of CSC surface markers (ALDH1 and CD44), and the capacity of sphereforming (mammospheres). We found that antagonism of miR-21 reversed EMT and CSC phenotype, accompanied with PTEN up-regulation and AKT/ERK1/2 inactivation. Interestingly, down-regulation of PTEN by siPTEN suppressed the effects of miR-21 antagomir on EMT and CSC phenotype, confirming that PTEN is a target of miR-21 in reversing EMT and CSC phenotype. The inhibitors of PI3K-AKT and ERK1/2 pathways, LY294002 and U0126, both significantly suppressed EMT and CSC phenotype, indicating that AKT and ERK1/2 pathways are required for miR-21 mediating EMT and CSC phenotype.
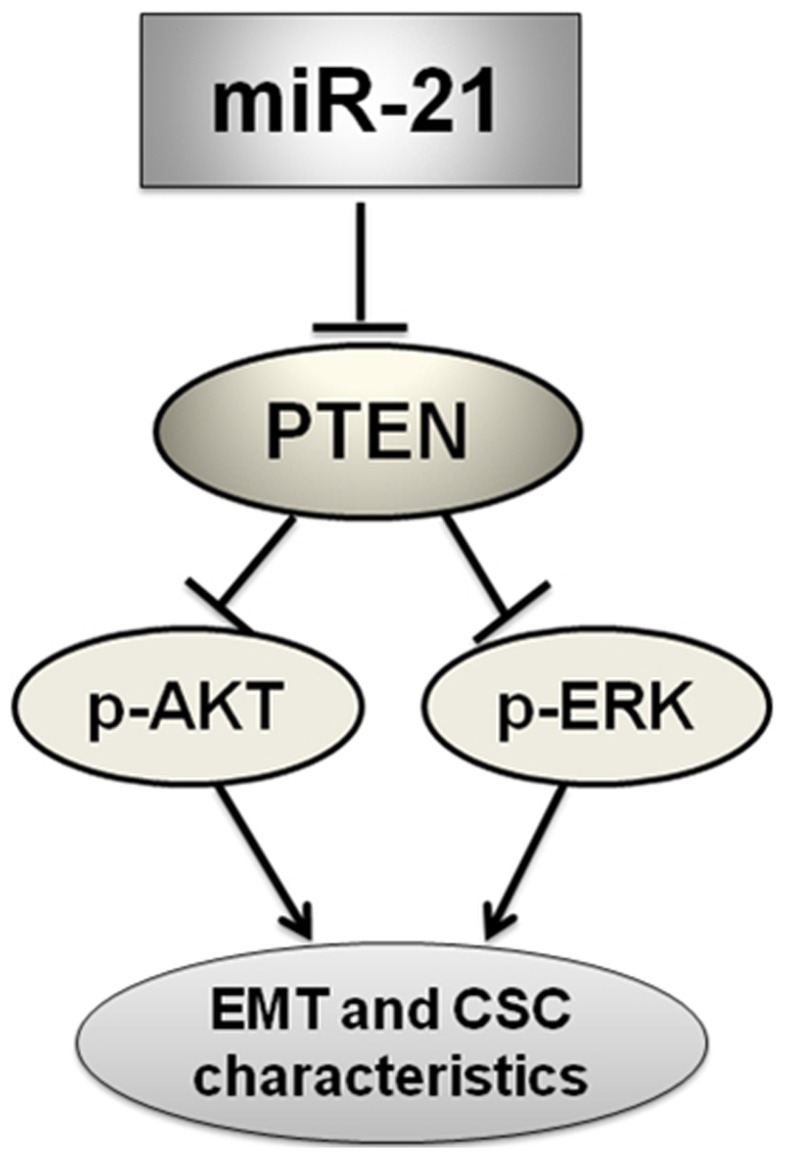
Conclusions/significance: In conclusion, our results demonstrated that antagonism of miR-21 reverses EMT and CSC phenotype through targeting PTEN, via inactivation of AKT and ERK1/2 pathways, and showed a novel mechanism of which might relieve the malignant biological behaviors of breast cancer.
Conflict of interest statement
Figures
References
-
- Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–454. - PubMed
-
- Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7:131–142. - PubMed
-
- Christiansen JJ, Rajasekaran AK. Reassessing epithelial to mesenchymal transition as a prerequisite for carcinoma invasion and metastasis. Cancer Res. 2006;66:8319–8326. - PubMed
-
- Thiery JP. Epithelial–mesenchymal transitions in development and pathologies. Curr Opin Cell Biol. 2000;15:740–746. - PubMed
-
- Polakis P. Wnt signaling and cancer. Genes Dev. 2000;14:1837–1851. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
