

The epoxyketone-based proteasome inhibitors carfilzomib and orally bioavailable oprozomib have anti-resorptive and bone-anabolic activity in addition to anti-myeloma effects
- PMID: 22763387
- PMCID: PMC3771507
- DOI: 10.1038/leu.2012.183
The epoxyketone-based proteasome inhibitors carfilzomib and orally bioavailable oprozomib have anti-resorptive and bone-anabolic activity in addition to anti-myeloma effects
Abstract
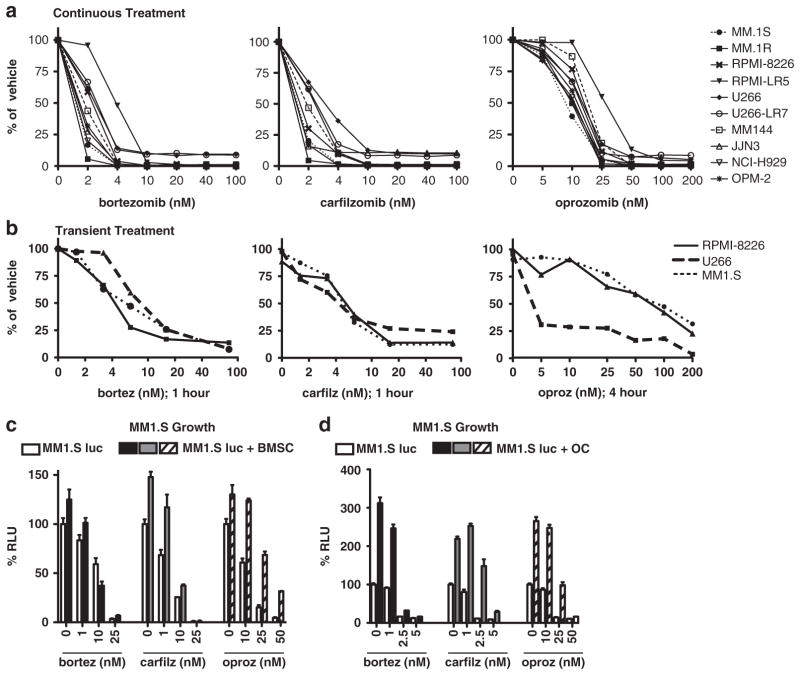
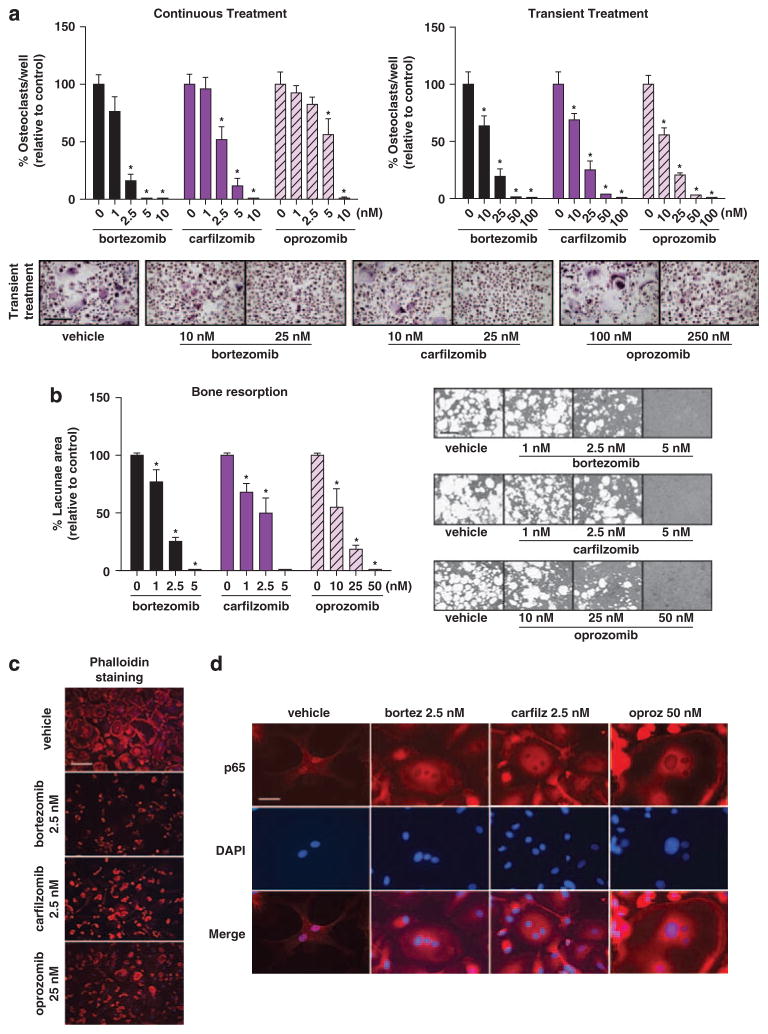
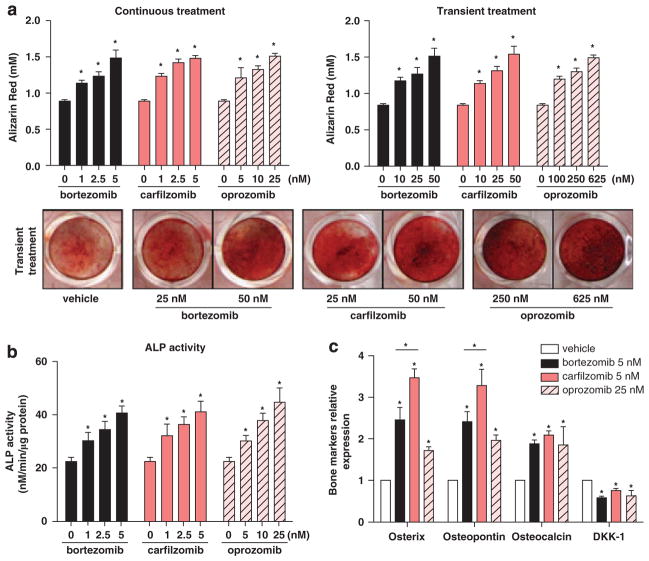
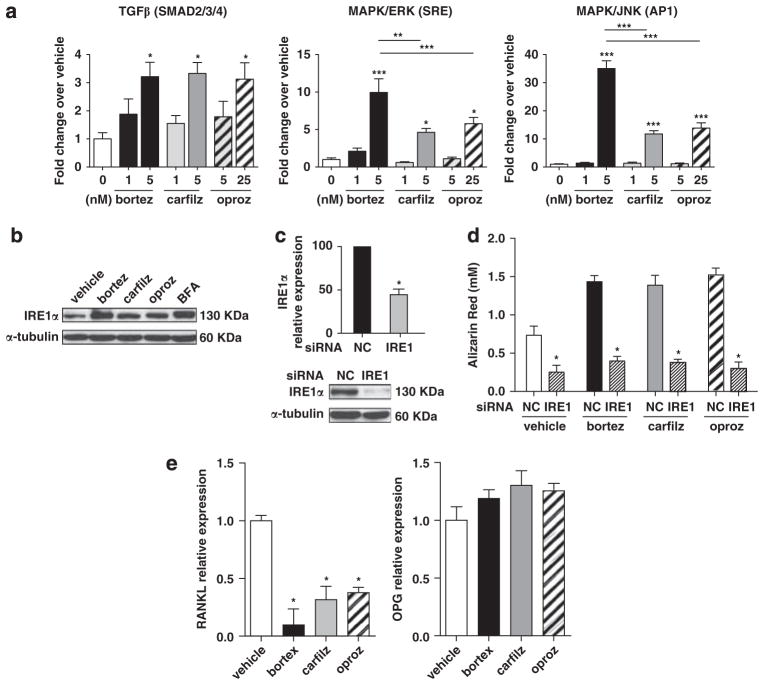
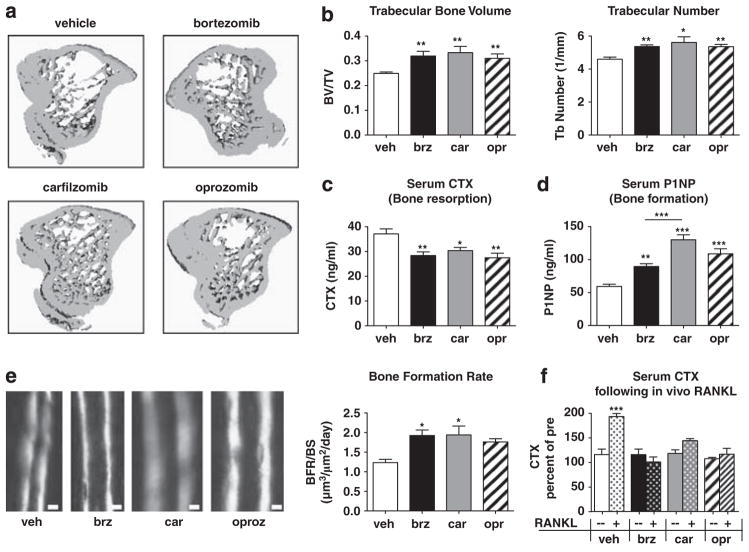
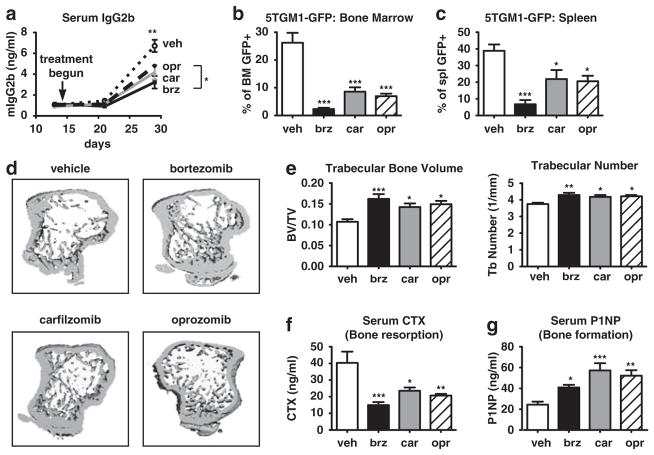
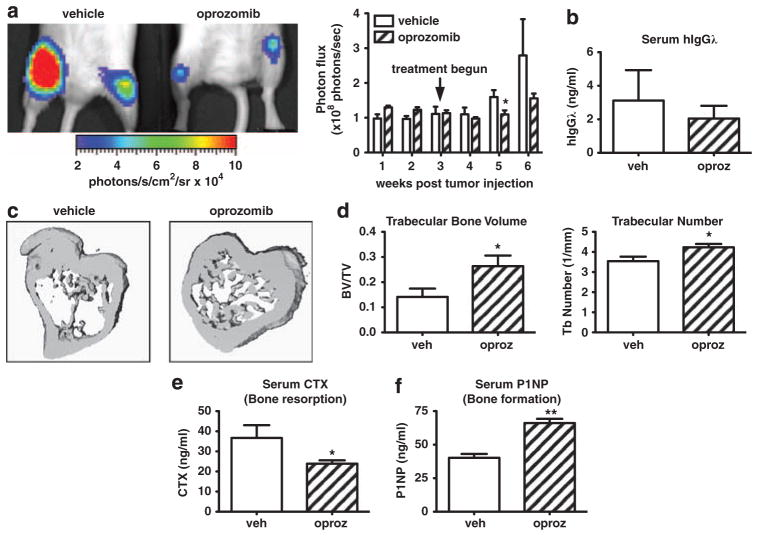
Proteasome inhibitors (PIs), namely bortezomib, have become a cornerstone therapy for multiple myeloma (MM), potently reducing tumor burden and inhibiting pathologic bone destruction. In clinical trials, carfilzomib, a next generation epoxyketone-based irreversible PI, has exhibited potent anti-myeloma efficacy and decreased side effects compared with bortezomib. Carfilzomib and its orally bioavailable analog oprozomib, effectively decreased MM cell viability following continual or transient treatment mimicking in vivo pharmacokinetics. Interactions between myeloma cells and the bone marrow (BM) microenvironment augment the number and activity of bone-resorbing osteoclasts (OCs) while inhibiting bone-forming osteoblasts (OBs), resulting in increased tumor growth and osteolytic lesions. At clinically relevant concentrations, carfilzomib and oprozomib directly inhibited OC formation and bone resorption in vitro, while enhancing osteogenic differentiation and matrix mineralization. Accordingly, carfilzomib and oprozomib increased trabecular bone volume, decreased bone resorption and enhanced bone formation in non-tumor bearing mice. Finally, in mouse models of disseminated MM, the epoxyketone-based PIs decreased murine 5TGM1 and human RPMI-8226 tumor burden and prevented bone loss. These data demonstrate that, in addition to anti-myeloma properties, carfilzomib and oprozomib effectively shift the bone microenvironment from a catabolic to an anabolic state and, similar to bortezomib, may decrease skeletal complications of MM.
Conflict of interest statement
CJK is an employee of Onyx Pharmaceuticals. All other authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
