

Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors
- PMID: 22763448
- PMCID: PMC3724525
- DOI: 10.1038/nature11249
Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors
Abstract
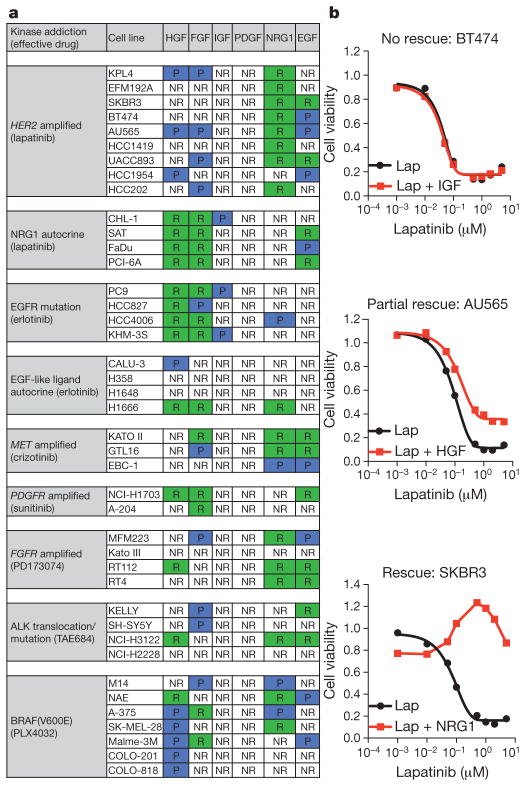
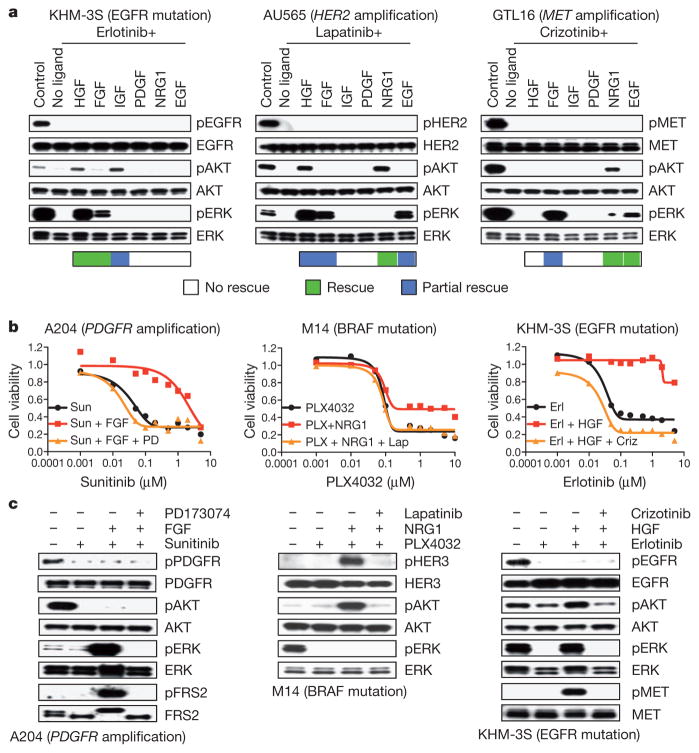
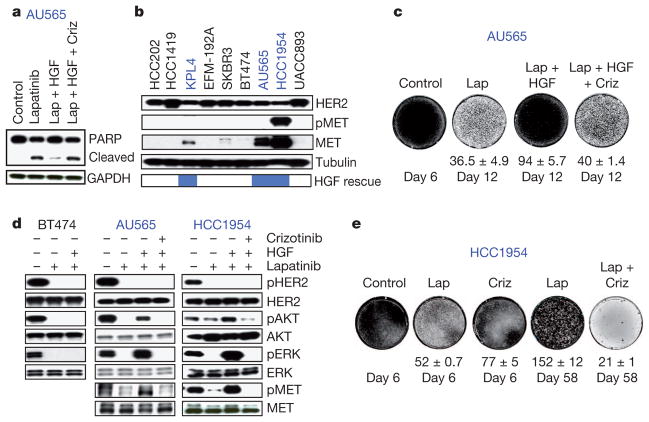
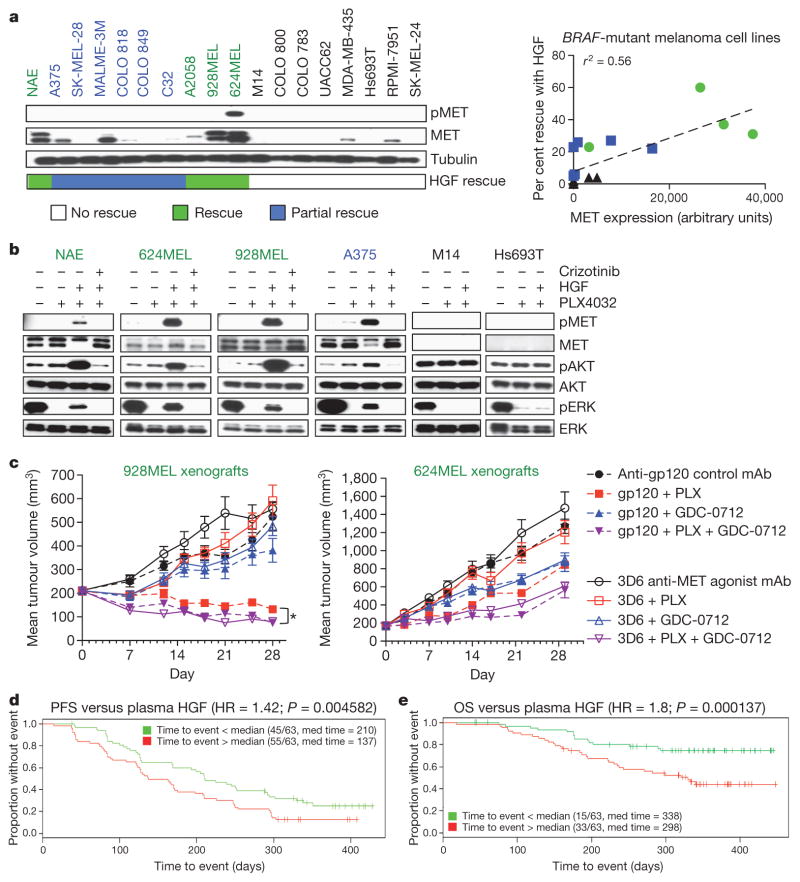
Mutationally activated kinases define a clinically validated class of targets for cancer drug therapy. However, the efficacy of kinase inhibitors in patients whose tumours harbour such alleles is invariably limited by innate or acquired drug resistance. The identification of resistance mechanisms has revealed a recurrent theme—the engagement of survival signals redundant to those transduced by the targeted kinase. Cancer cells typically express multiple receptor tyrosine kinases (RTKs) that mediate signals that converge on common critical downstream cell-survival effectors—most notably, phosphatidylinositol-3-OH kinase (PI(3)K) and mitogen-activated protein kinase (MAPK). Consequently, an increase in RTK-ligand levels, through autocrine tumour-cell production, paracrine contribution from tumour stroma or systemic production, could confer resistance to inhibitors of an oncogenic kinase with a similar signalling output. Here, using a panel of kinase-'addicted' human cancer cell lines, we found that most cells can be rescued from drug sensitivity by simply exposing them to one or more RTK ligands. Among the findings with clinical implications was the observation that hepatocyte growth factor (HGF) confers resistance to the BRAF inhibitor PLX4032 (vemurafenib) in BRAF-mutant melanoma cells. These observations highlight the extensive redundancy of RTK-transduced signalling in cancer cells and the potentially broad role of widely expressed RTK ligands in innate and acquired resistance to drugs targeting oncogenic kinases.
Figures
Comment in
-
Targeted therapies: hepatocyte growth factor-a culprit of drug resistance.Nat Rev Clin Oncol. 2012 Jul 17;9(8):429. doi: 10.1038/nrclinonc.2012.124. Nat Rev Clin Oncol. 2012. PMID: 22801668 No abstract available.
-
The haves and the have nots.Nat Rev Cancer. 2012 Jul 24;12(8):505. doi: 10.1038/nrc3330. Nat Rev Cancer. 2012. PMID: 22825209 No abstract available.
-
Registered report: Widespread potential for growth factor-driven resistance to anticancer kinase inhibitors.Elife. 2014 Dec 10;3:e04037. doi: 10.7554/eLife.04037. Elife. 2014. PMID: 25490934 Free PMC article.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
