

Saturated fatty acids activate TLR-mediated proinflammatory signaling pathways
- PMID: 22766885
- PMCID: PMC3413240
- DOI: 10.1194/jlr.D029546
Saturated fatty acids activate TLR-mediated proinflammatory signaling pathways
Abstract
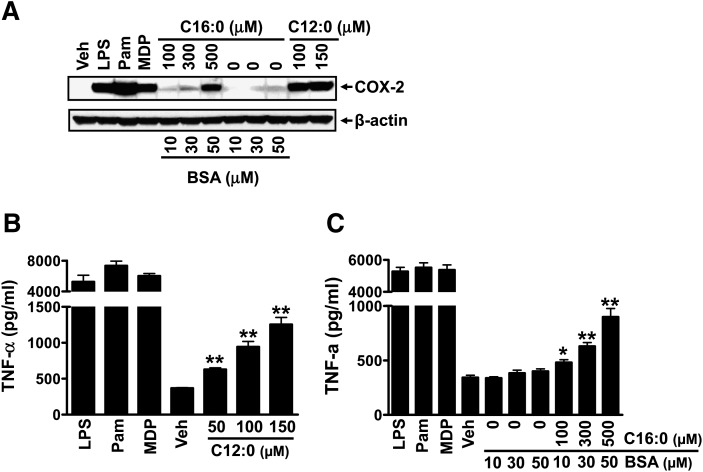
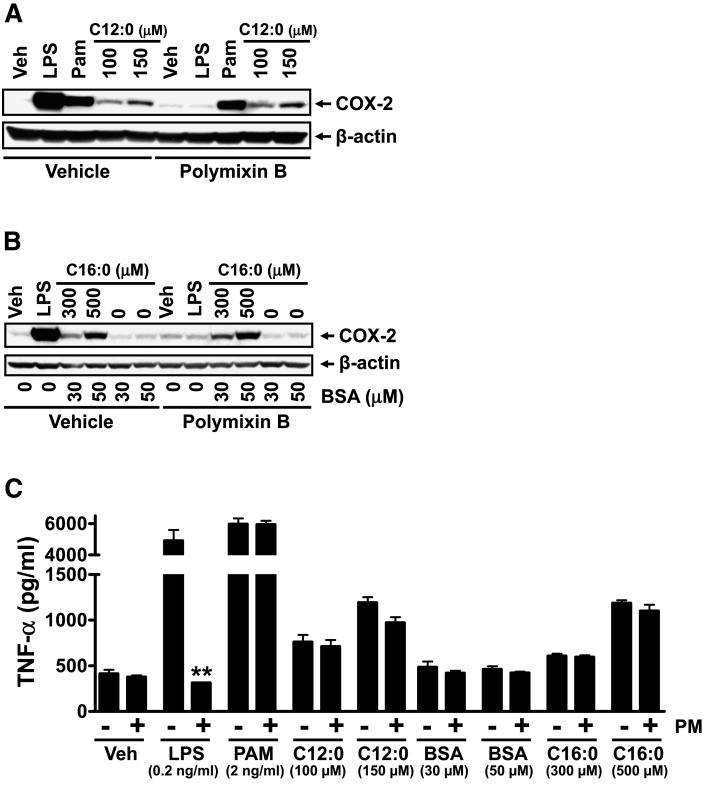
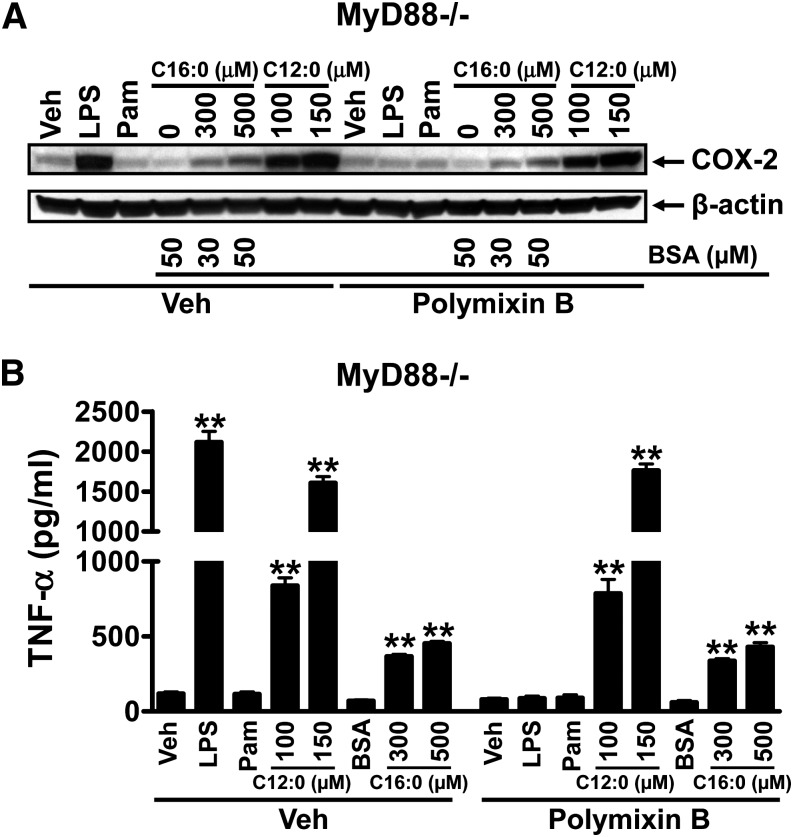
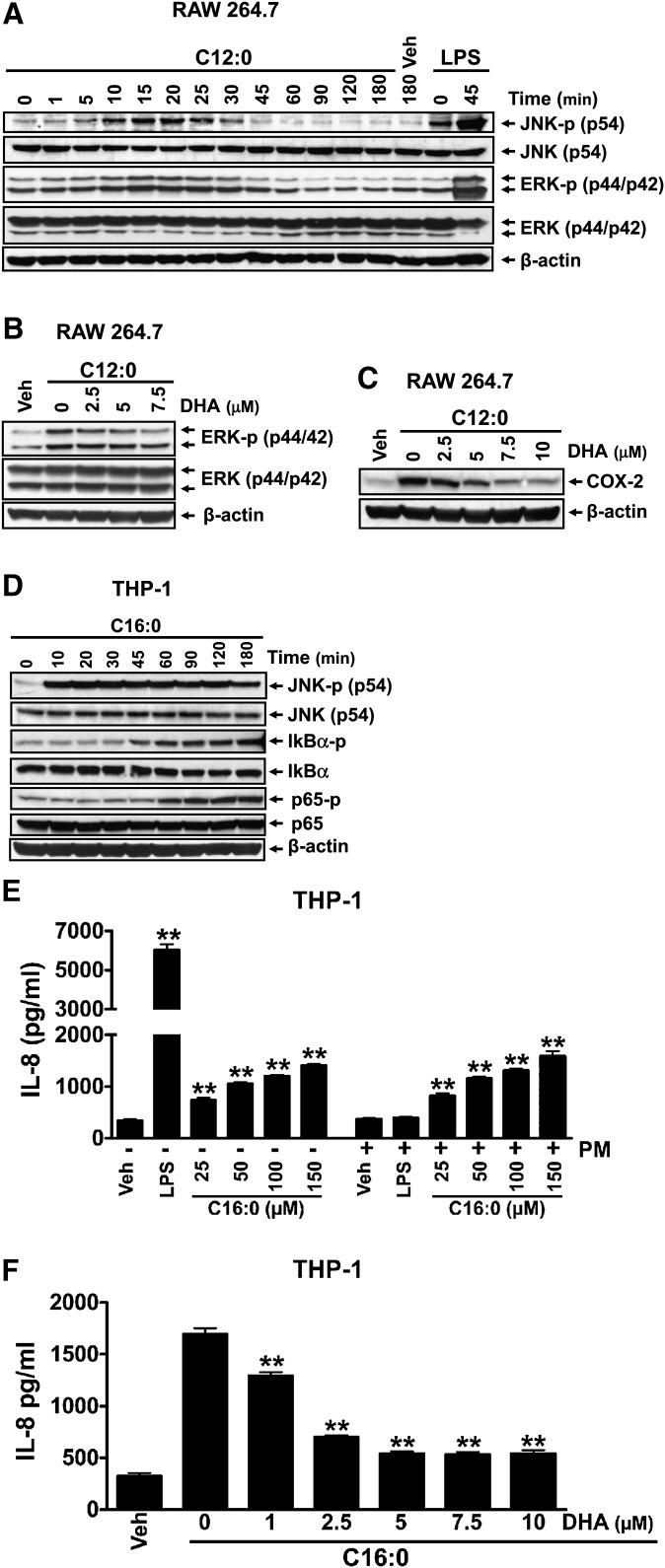
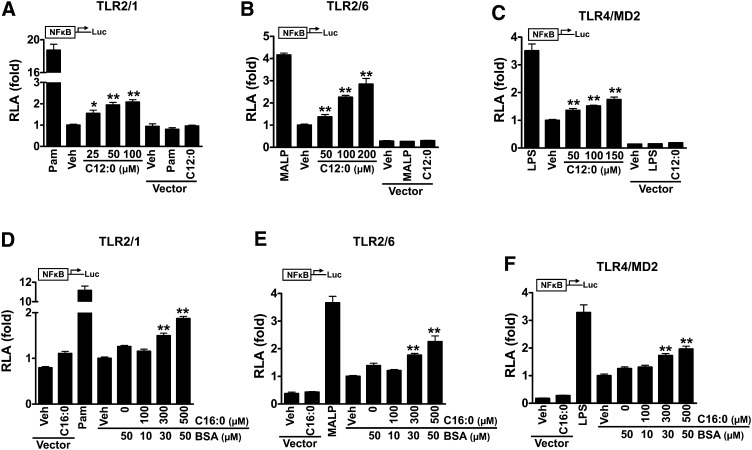
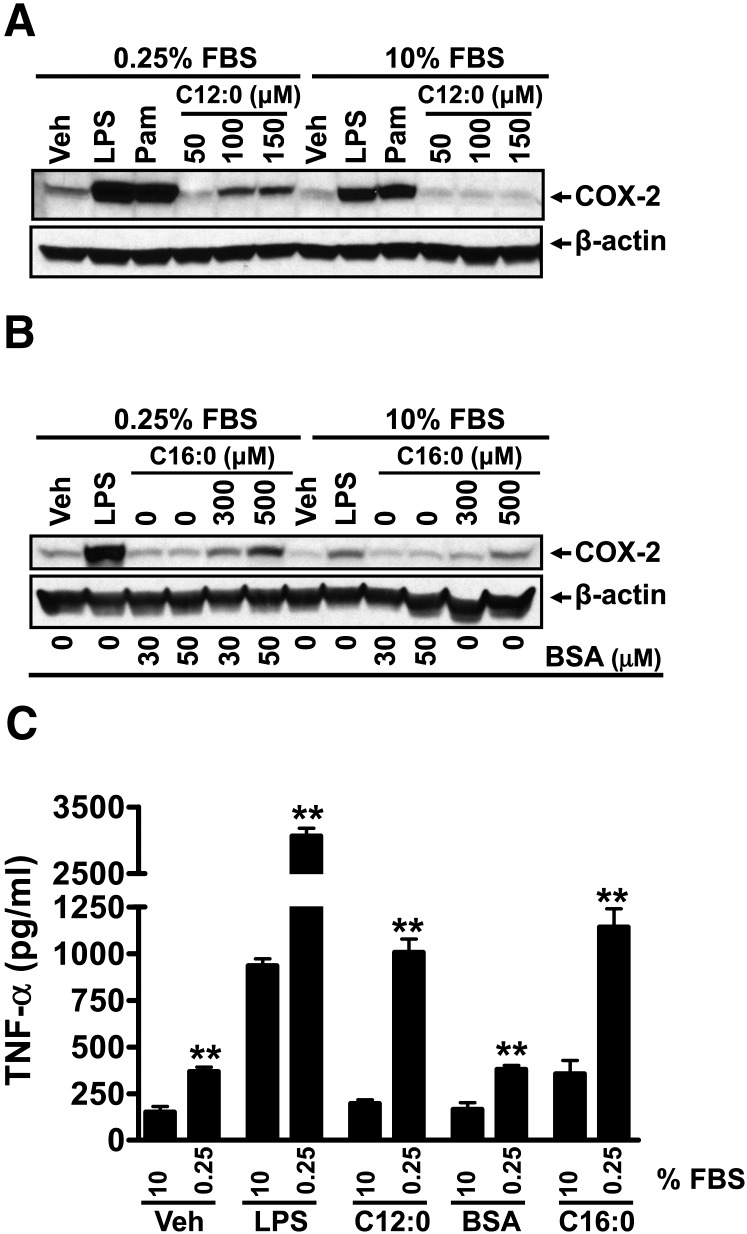
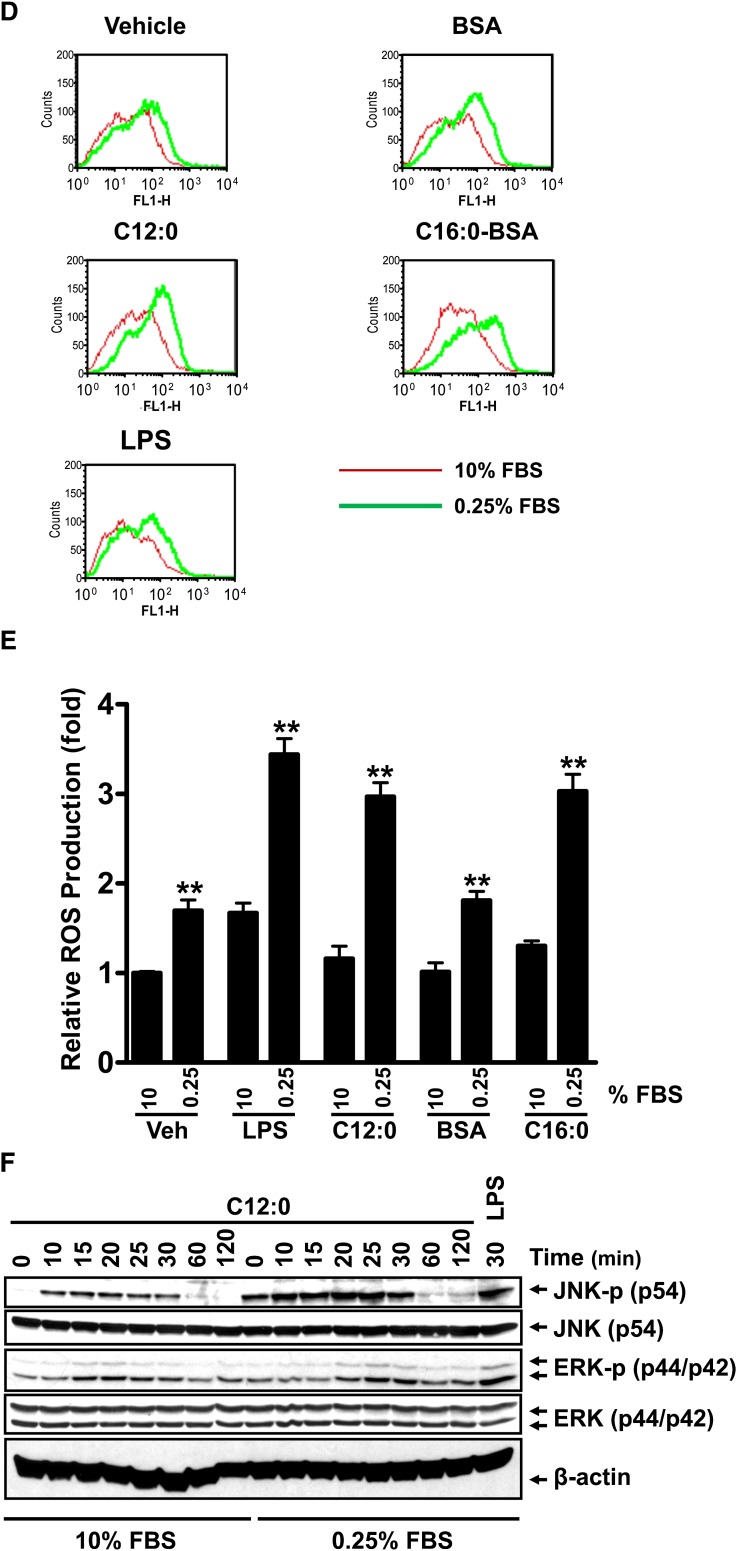
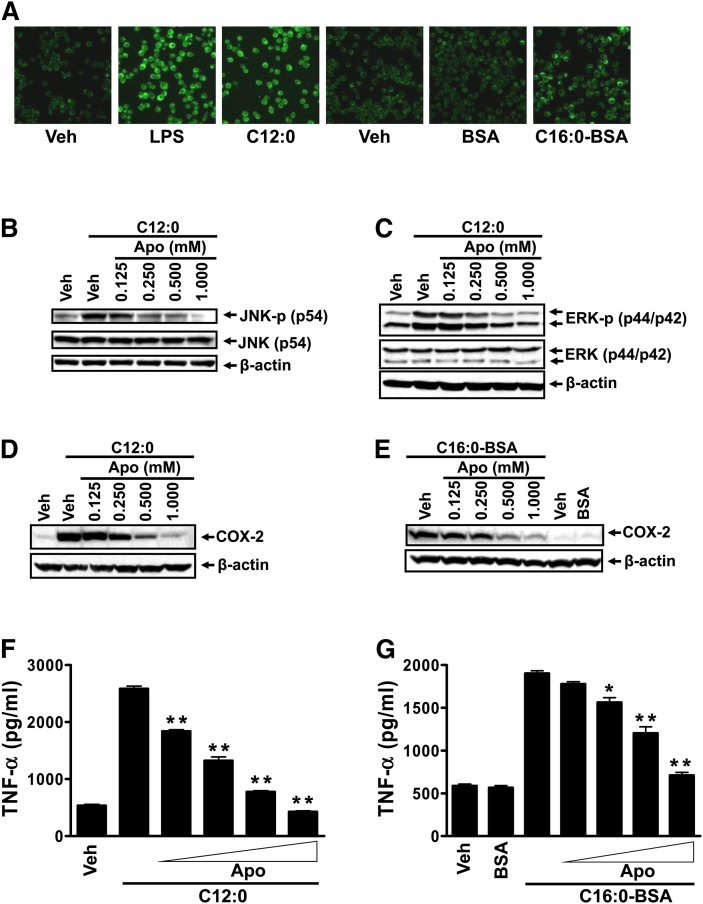
Toll-like receptor 4 (TLR4) and TLR2 were shown to be activated by saturated fatty acids (SFAs) but inhibited by docosahexaenoic acid (DHA). However, one report suggested that SFA-induced TLR activation in cell culture systems is due to contaminants in BSA used for solubilizing fatty acids. This report raised doubt about proinflammatory effects of SFAs. Our studies herein demonstrate that sodium palmitate (C16:0) or laurate (C12:0) without BSA solubilization induced phosphorylation of inhibitor of nuclear factor-κB α, c-Jun N-terminal kinase (JNK), p44/42 mitogen-activated-kinase (ERK), and nuclear factor-κB subunit p65, and TLR target gene expression in THP1 monocytes or RAW264.7 macrophages, respectively, when cultured in low FBS (0.25%) medium. C12:0 induced NFκB activation through TLR2 dimerized with TLR1 or TLR6, and through TLR4. Because BSA was not used in these experiments, contaminants in BSA have no relevance. Unlike in suspension cells (THP-1), BSA-solubilized C16:0 instead of sodium C16:0 is required to induce TLR target gene expression in adherent cells (RAW264.7). C16:0-BSA transactivated TLR2 dimerized with TLR1 or TLR6 and through TLR4 as seen with C12:0. These results and additional studies with the LPS sequester polymixin B and in MyD88(-/-) macrophages indicated that SFA-induced activation of TLR2 or TLR4 is a fatty acid-specific effect, but not due to contaminants in BSA or fatty acid preparations.
Figures
References
-
- Schroder K., Zhou R., Tschopp J. 2010. The NLRP3 inflammasome: a sensor for metabolic danger? Science. 327: 296–300. - PubMed
-
- Lee J. Y., Zhao L., Hwang D. H. 2010. Modulation of pattern recognition receptor-mediated inflammation and risk of chronic diseases by dietary fatty acids. Nutr. Rev. 68: 38–61. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
