

Probing of exosites leads to novel inhibitor scaffolds of HCV NS3/4A proteinase
- PMID: 22768327
- PMCID: PMC3388044
- DOI: 10.1371/journal.pone.0040029
Probing of exosites leads to novel inhibitor scaffolds of HCV NS3/4A proteinase
Abstract
Background: Hepatitis C is a treatment-resistant disease affecting millions of people worldwide. The hepatitis C virus (HCV) genome is a single-stranded RNA molecule. After infection of the host cell, viral RNA is translated into a polyprotein that is cleaved by host and viral proteinases into functional, structural and non-structural, viral proteins. Cleavage of the polyprotein involves the viral NS3/4A proteinase, a proven drug target. HCV mutates as it replicates and, as a result, multiple emerging quasispecies become rapidly resistant to anti-virals, including NS3/4A inhibitors.
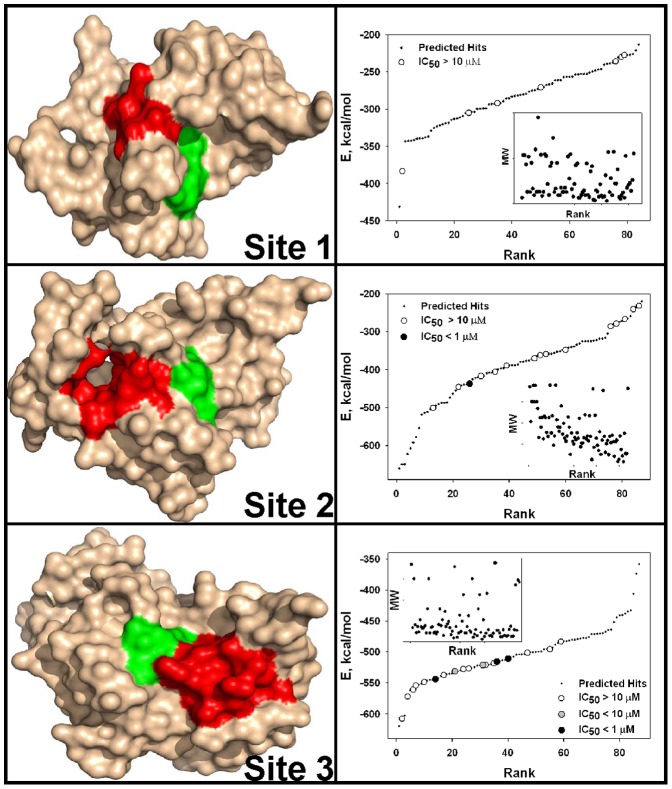
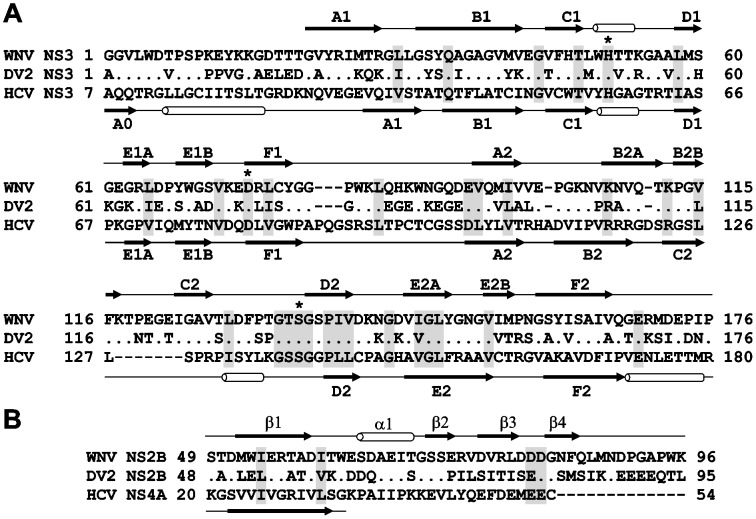
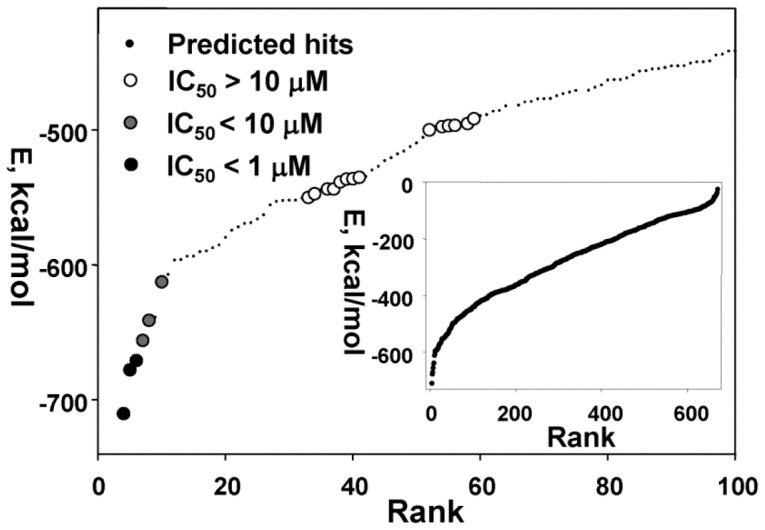
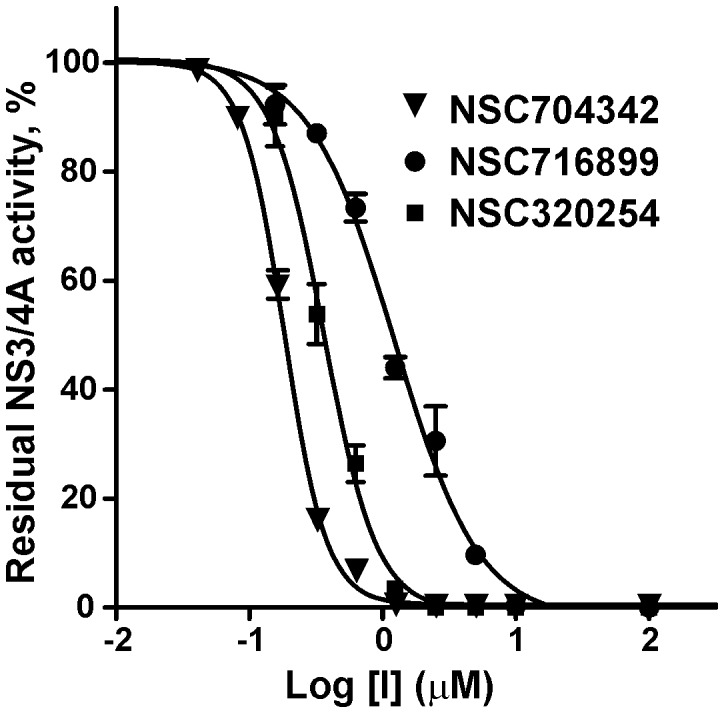
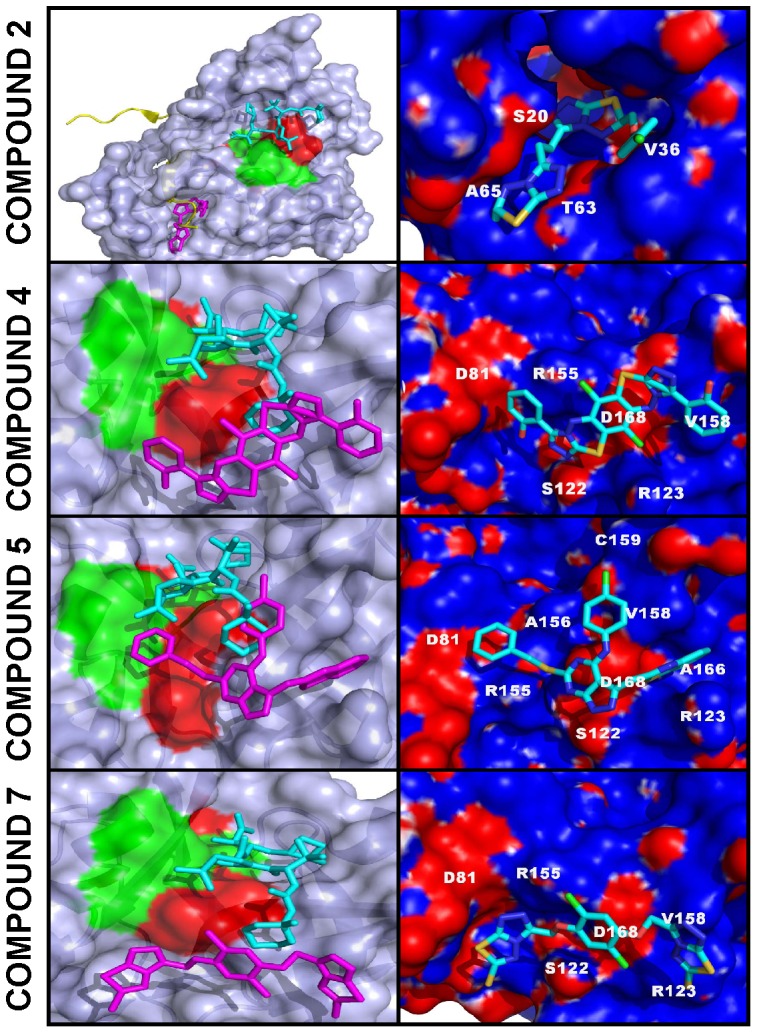
Methodology/principal findings: To circumvent drug resistance and complement the existing anti-virals, NS3/4A inhibitors, which are additional and distinct from the FDA-approved telaprevir and boceprevir α-ketoamide inhibitors, are required. To test potential new avenues for inhibitor development, we have probed several distinct exosites of NS3/4A which are either outside of or partially overlapping with the active site groove of the proteinase. For this purpose, we employed virtual ligand screening using the 275,000 compound library of the Developmental Therapeutics Program (NCI/NIH) and the X-ray crystal structure of NS3/4A as a ligand source and a target, respectively. As a result, we identified several novel, previously uncharacterized, nanomolar range inhibitory scaffolds, which suppressed of the NS3/4A activity in vitro and replication of a sub-genomic HCV RNA replicon with a luciferase reporter in human hepatocarcinoma cells. The binding sites of these novel inhibitors do not significantly overlap with those of α-ketoamides. As a result, the most common resistant mutations, including V36M, R155K, A156T, D168A and V170A, did not considerably diminish the inhibitory potency of certain novel inhibitor scaffolds we identified.
Conclusions/significance: Overall, the further optimization of both the in silico strategy and software platform we developed and lead compounds we identified may lead to advances in novel anti-virals.
Conflict of interest statement
Figures
References
-
- Chevaliez S, Pawlotsky JM. HCV genome and life cycle. In: Tan SL, editor. Chapter 1 Hepatitis C viruses: genomes and molecular biology. Norfolk (UK): Horizon Bioscience; 2006. pp. 5–47. - PubMed
-
- Penin F, Dubuisson J, Rey FA, Moradpour D, Pawlotsky JM. Structural biology of hepatitis C virus. Hepatology. 2004;39:5–19. - PubMed
-
- Kim JL, Morgenstern KA, Lin C, Fox T, Dwyer MD, et al. Crystal structure of the hepatitis C virus NS3 protease domain complexed with a synthetic NS4A cofactor peptide. Cell. 1996;87:343–355. - PubMed
-
- Kwong AD, Kim JL, Rao G, Lipovsek D, Raybuck SA. Hepatitis C virus NS3/4A protease. Antiviral Res. 1999;41:67–84. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
