

Complete genome sequence of Enterococcus faecium strain TX16 and comparative genomic analysis of Enterococcus faecium genomes
- PMID: 22769602
- PMCID: PMC3433357
- DOI: 10.1186/1471-2180-12-135
Complete genome sequence of Enterococcus faecium strain TX16 and comparative genomic analysis of Enterococcus faecium genomes
Abstract
Background: Enterococci are among the leading causes of hospital-acquired infections in the United States and Europe, with Enterococcus faecalis and Enterococcus faecium being the two most common species isolated from enterococcal infections. In the last decade, the proportion of enterococcal infections caused by E. faecium has steadily increased compared to other Enterococcus species. Although the underlying mechanism for the gradual replacement of E. faecalis by E. faecium in the hospital environment is not yet understood, many studies using genotyping and phylogenetic analysis have shown the emergence of a globally dispersed polyclonal subcluster of E. faecium strains in clinical environments. Systematic study of the molecular epidemiology and pathogenesis of E. faecium has been hindered by the lack of closed, complete E. faecium genomes that can be used as references.
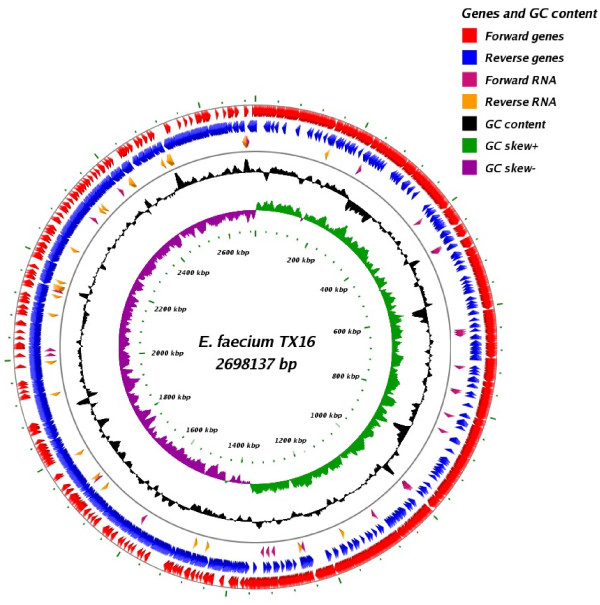
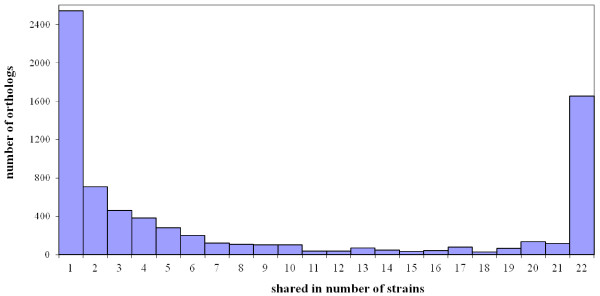
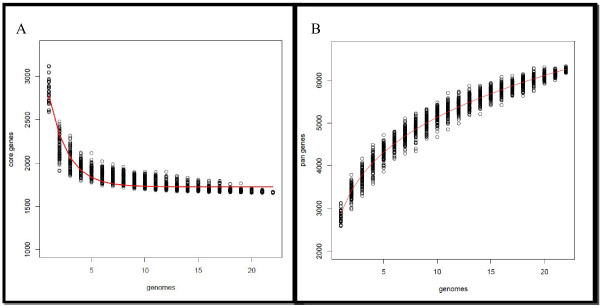
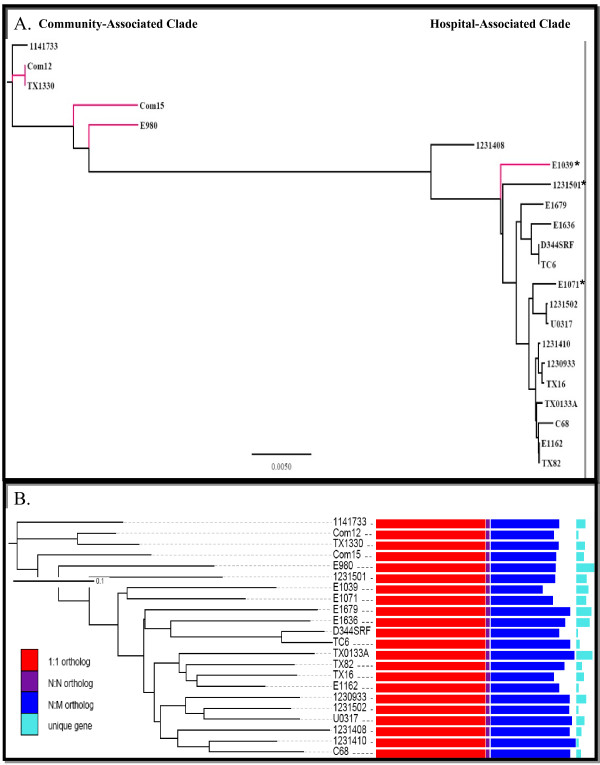
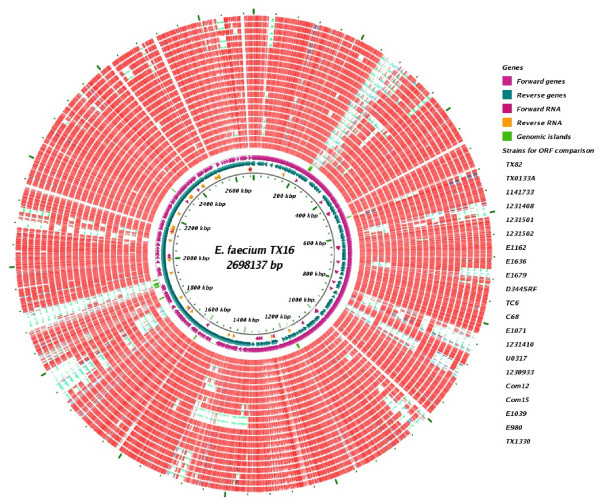
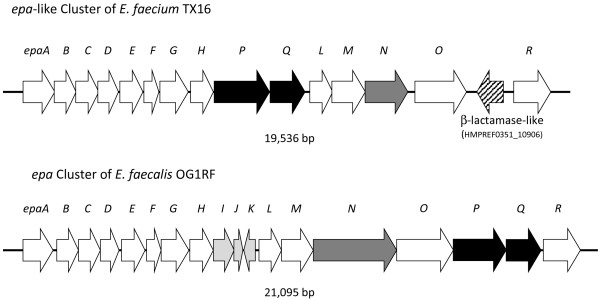
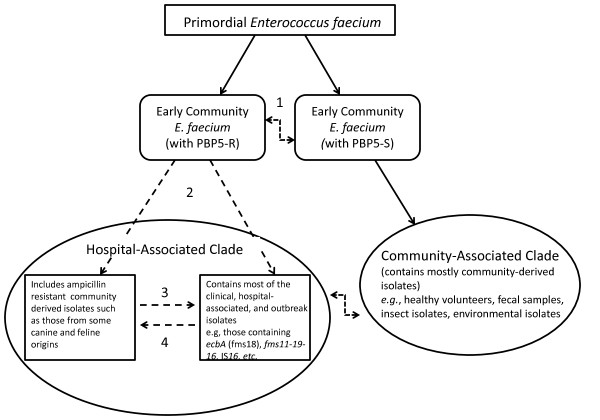
Results: In this study, we report the complete genome sequence of the E. faecium strain TX16, also known as DO, which belongs to multilocus sequence type (ST) 18, and was the first E. faecium strain ever sequenced. Whole genome comparison of the TX16 genome with 21 E. faecium draft genomes confirmed that most clinical, outbreak, and hospital-associated (HA) strains (including STs 16, 17, 18, and 78), in addition to strains of non-hospital origin, group in the same clade (referred to as the HA clade) and are evolutionally considerably more closely related to each other by phylogenetic and gene content similarity analyses than to isolates in the community-associated (CA) clade with approximately a 3-4% average nucleotide sequence difference between the two clades at the core genome level. Our study also revealed that many genomic loci in the TX16 genome are unique to the HA clade. 380 ORFs in TX16 are HA-clade specific and antibiotic resistance genes are enriched in HA-clade strains. Mobile elements such as IS16 and transposons were also found almost exclusively in HA strains, as previously reported.
Conclusions: Our findings along with other studies show that HA clonal lineages harbor specific genetic elements as well as sequence differences in the core genome which may confer selection advantages over the more heterogeneous CA E. faecium isolates. Which of these differences are important for the success of specific E. faecium lineages in the hospital environment remain(s) to be determined.
Figures
References
-
- Willems RJ, van Schaik W. Transition of Enterococcus faecium from commensal organism to nosocomial pathogen. Future Microbiol. 2009;4(9):1125–1135. - PubMed
-
- Hidron AI, Edwards JR, Patel J, Horan TC, Sievert DM, Pollock DA, Fridkin SK. NHSN annual update: antimicrobial-resistant pathogens associated with healthcare-associated infections: annual summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2006–2007. Infect Control Hosp Epidemiol. 2008;29(11):996–1011. - PubMed
-
- Leavis HL, Bonten MJ, Willems RJ. Identification of high-risk enterococcal clonal complexes: global dispersion and antibiotic resistance. Curr Opin Microbiol. 2006;9(5):454–460. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
