

Association with nitric oxide synthase on insulin secretory granules regulates glucokinase protein levels
- PMID: 22771492
- PMCID: PMC3434526
- DOI: 10.1210/me.2012-1183
Association with nitric oxide synthase on insulin secretory granules regulates glucokinase protein levels
Abstract
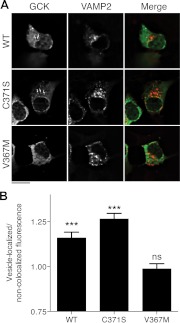
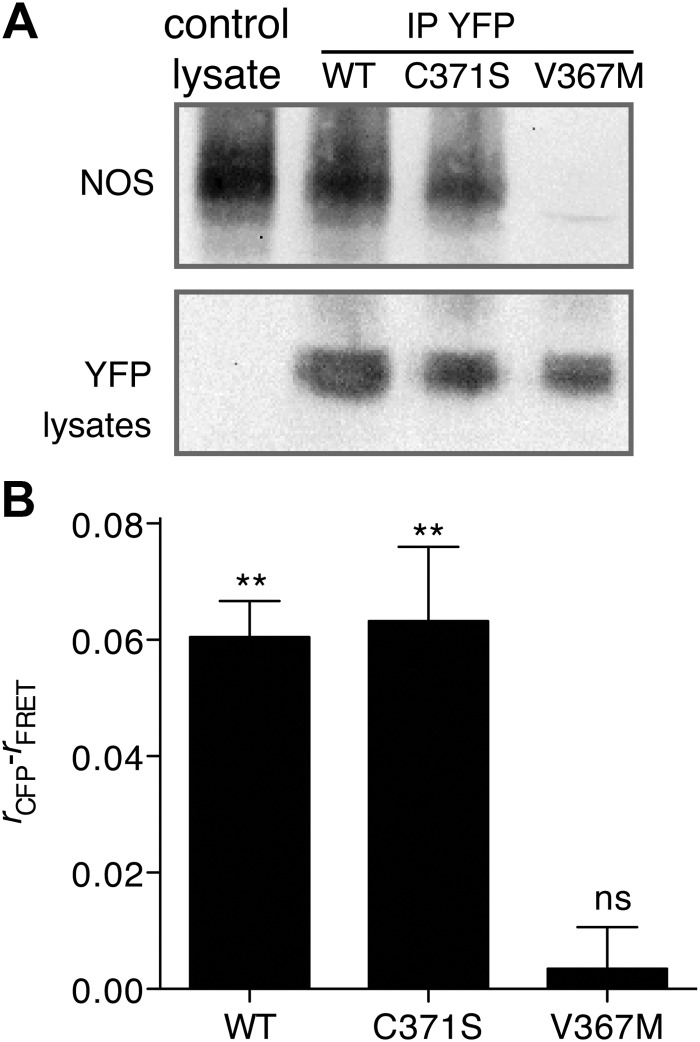
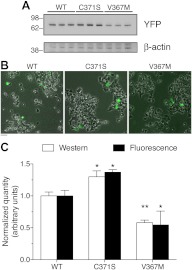
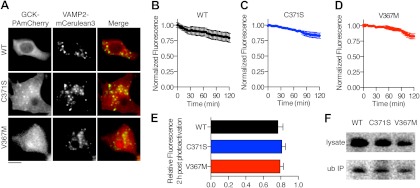
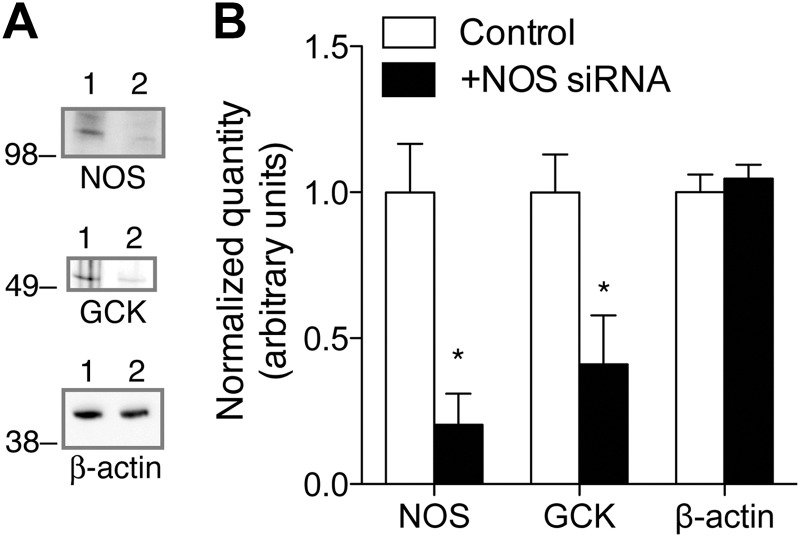
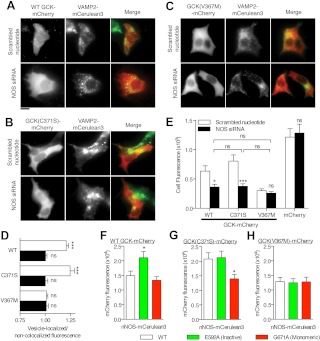
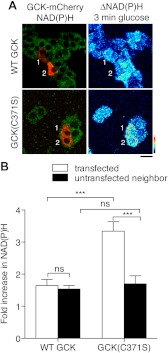
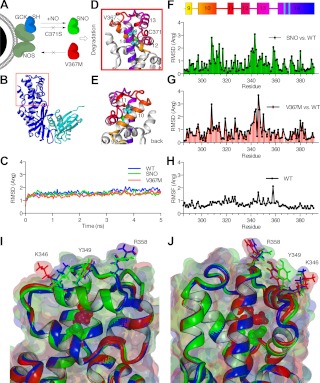
Glucokinase (GCK) association with insulin-secretory granules is controlled by interaction with nitric oxide synthase (NOS) and is reversed by GCK S-nitrosylation. Nonetheless, the function of GCK sequestration on secretory granules is unknown. Here we report that the S-nitrosylation blocking V367M mutation prevents GCK accumulation on secretory granules by inhibiting association with NOS. Expression of this mutant is reduced compared with a second S-nitrosylation blocking GCK mutant (C371S) that accumulates to secretory granules and is expressed at levels greater than wild type. Even so, the rate of degradation for wild type and mutant GCK proteins were not significantly different from one another, and neither mutation disrupted the ability of GCK to be ubiquitinated. Furthermore, gene silencing of NOS reduced endogenous GCK content but did not affect β-actin content. Treatment of GCK(C371S) expressing cells with short interfering RNA specific for NOS also blocked accumulation of this protein to secretory granules and reduced expression levels to that of GCK(V367M). Conversely, cotransfection of catalytically inactive NOS increased GCK-mCherry levels. Expression of GCK(C371S) in βTC3 cells enhanced glucose metabolism compared with untransfected cells and cells expressing wild type GCK, even though this mutant has slightly reduced enzymatic activity in vitro. Finally, molecular dynamics simulations revealed that V367M induces conformational changes in GCK that are similar to S-nitrosylated GCK, thereby suggesting a mechanism for V367M-inhibition of NOS association. Our findings suggest that sequestration of GCK on secretory granules regulates cellular GCK protein content, and thus cellular GCK activity, by acting as a storage pool for GCK proteins.
Figures
Similar articles
-
Molecular and cellular regulation of human glucokinase.Arch Biochem Biophys. 2019 Mar 15;663:199-213. doi: 10.1016/j.abb.2019.01.011. Epub 2019 Jan 11. Arch Biochem Biophys. 2019. PMID: 30641049 Free PMC article. Review.
-
Glucagon-like peptide 1 stimulates post-translational activation of glucokinase in pancreatic beta cells.J Biol Chem. 2011 May 13;286(19):16768-74. doi: 10.1074/jbc.M110.192799. Epub 2011 Mar 25. J Biol Chem. 2011. PMID: 21454584 Free PMC article.
-
Regulation of beta cell glucokinase by S-nitrosylation and association with nitric oxide synthase.J Cell Biol. 2003 Apr 28;161(2):243-8. doi: 10.1083/jcb.200301063. Epub 2003 Apr 21. J Cell Biol. 2003. PMID: 12707306 Free PMC article.
-
Naturally occurring glucokinase mutations are associated with defects in posttranslational S-nitrosylation.Mol Endocrinol. 2010 Jan;24(1):171-7. doi: 10.1210/me.2009-0138. Epub 2009 Nov 24. Mol Endocrinol. 2010. PMID: 19934346 Free PMC article.
-
A report of 2 new cases of MODY2 and review of the literature: implications in the search for type 2 diabetes drugs.Metabolism. 2013 Nov;62(11):1535-42. doi: 10.1016/j.metabol.2013.06.007. Epub 2013 Jul 24. Metabolism. 2013. PMID: 23890519 Review.
Cited by
-
Molecular and cellular regulation of human glucokinase.Arch Biochem Biophys. 2019 Mar 15;663:199-213. doi: 10.1016/j.abb.2019.01.011. Epub 2019 Jan 11. Arch Biochem Biophys. 2019. PMID: 30641049 Free PMC article. Review.
-
Identification of the ubiquitin-like domain of midnolin as a new glucokinase interaction partner.J Biol Chem. 2013 Dec 13;288(50):35824-39. doi: 10.1074/jbc.M113.526632. Epub 2013 Nov 1. J Biol Chem. 2013. PMID: 24187134 Free PMC article.
-
The manifold roles of protein S-nitrosylation in the life of insulin.Nat Rev Endocrinol. 2022 Feb;18(2):111-128. doi: 10.1038/s41574-021-00583-1. Epub 2021 Nov 17. Nat Rev Endocrinol. 2022. PMID: 34789923 Free PMC article. Review.
-
Protein posttranslational modifications in health and diseases: Functions, regulatory mechanisms, and therapeutic implications.MedComm (2020). 2023 May 2;4(3):e261. doi: 10.1002/mco2.261. eCollection 2023 Jun. MedComm (2020). 2023. PMID: 37143582 Free PMC article. Review.
-
Nitric Oxide Activates β-Cell Glucokinase by Promoting Formation of the "Glucose-Activated" State.Biochemistry. 2018 Aug 28;57(34):5136-5144. doi: 10.1021/acs.biochem.8b00333. Epub 2018 Aug 10. Biochemistry. 2018. PMID: 30053375 Free PMC article.
References
-
- Gloyn AL, Odili S, Buettger C, Njolstad PR, Shiota C, Magnuson MA, Matschinsky FM. 2004. Glucokinase and the regulation of blood sugar. In: Matschinsky FM, Magnuson MA, eds. Glucokinase and glycemic disease: from basics to novel therapeutics. Basel, Karger; 92–109
-
- Matschinsky FM. 2002. Regulation of pancreatic β-cell glucokinase: from basics to therapeutics. Diabetes 51( Suppl 3):S394–S404 - PubMed
-
- Davis EA, Cuesta-Muñoz A, Raoul M, Buettger C, Sweet I, Moates M, Magnuson MA, Matschinsky FM. 1999. Mutants of glucokinase cause hypoglycaemia- and hyperglycaemia syndromes and their analysis illuminates fundamental quantitative concepts of glucose homeostasis. Diabetologia 42:1175–1186 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
