

Mapping L1 ligase ribozyme conformational switch
- PMID: 22771572
- PMCID: PMC3509952
- DOI: 10.1016/j.jmb.2012.06.035
Mapping L1 ligase ribozyme conformational switch
Abstract
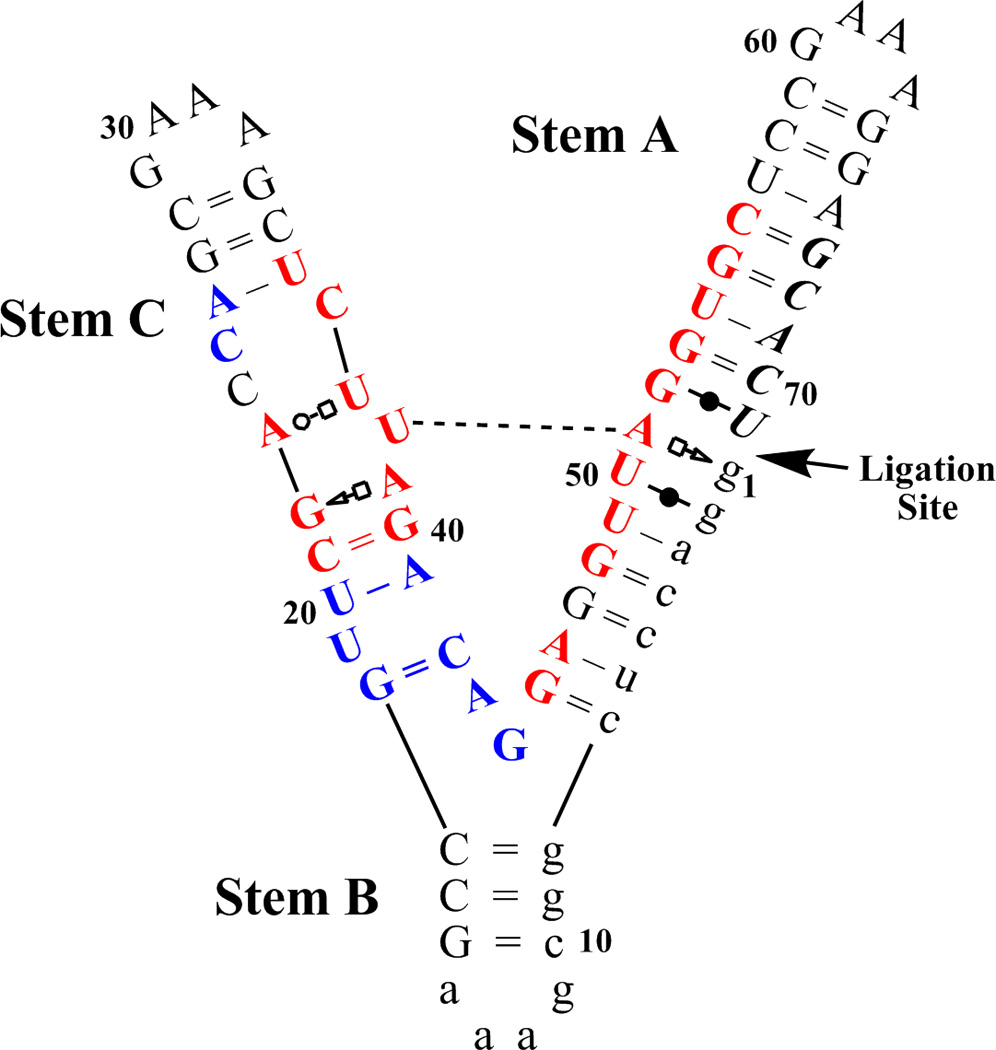
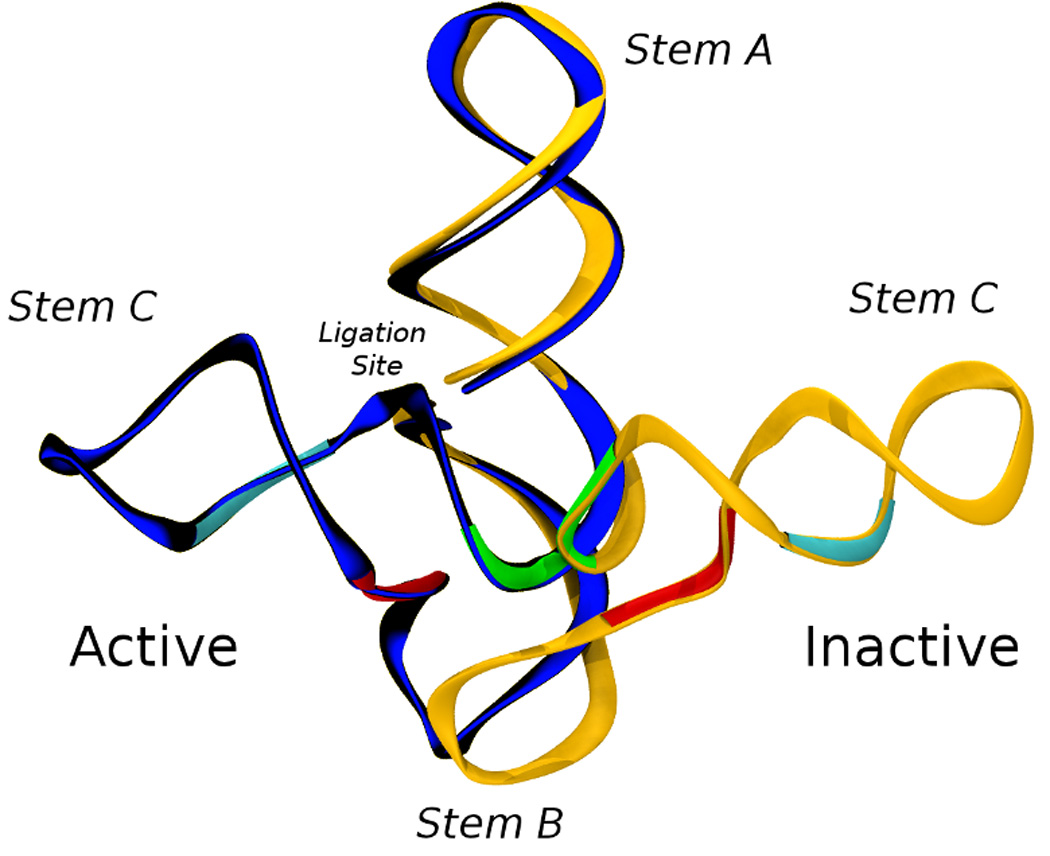
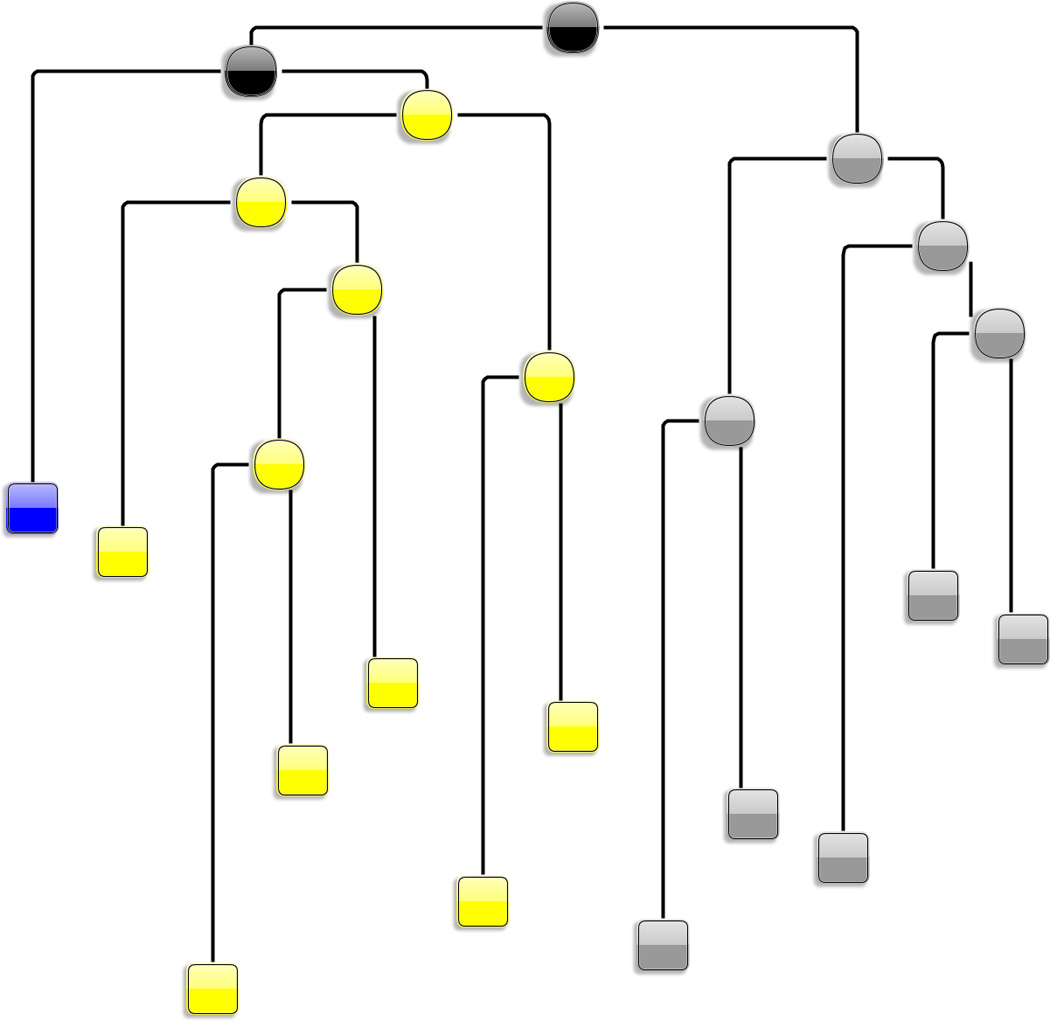
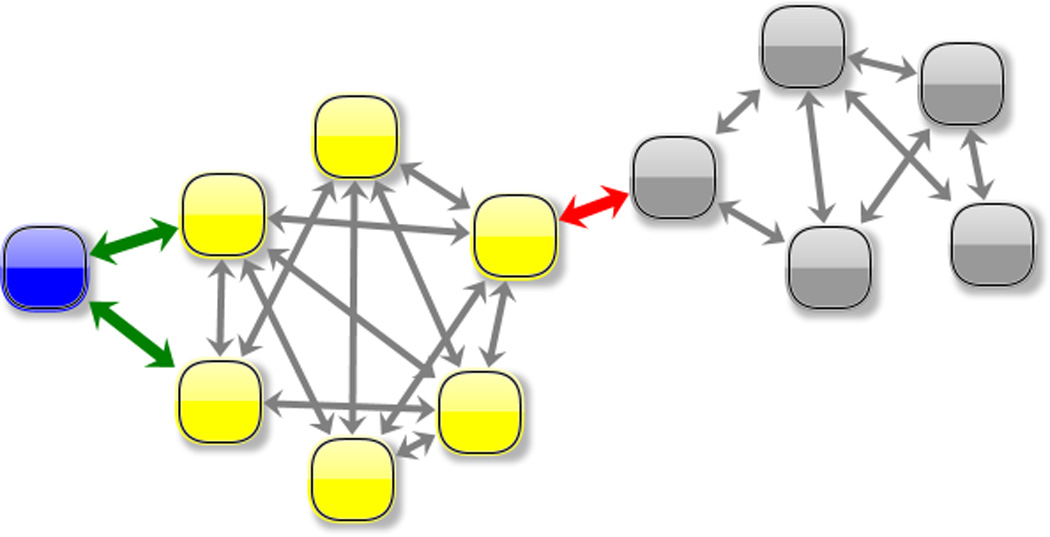
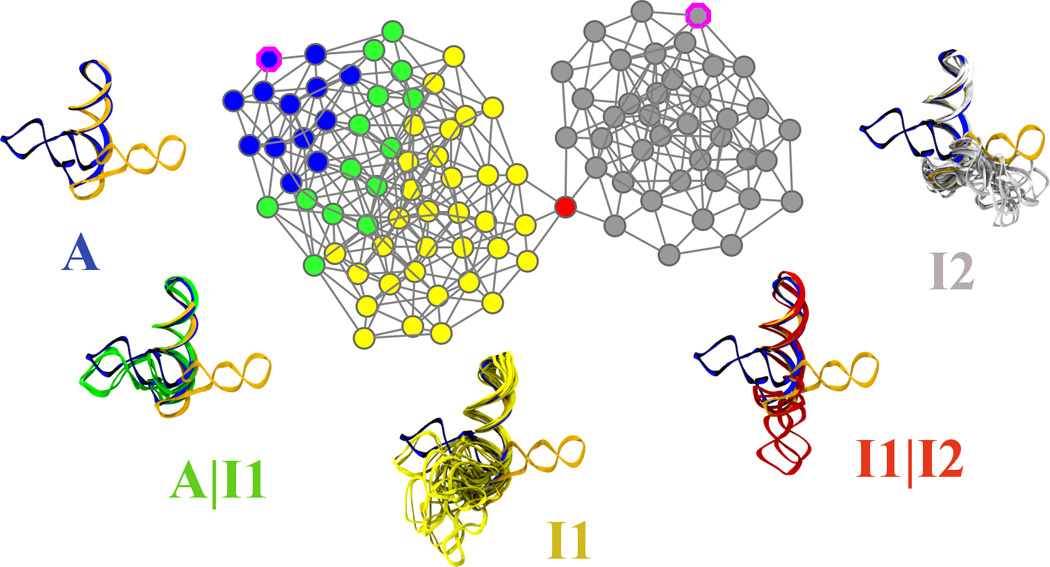
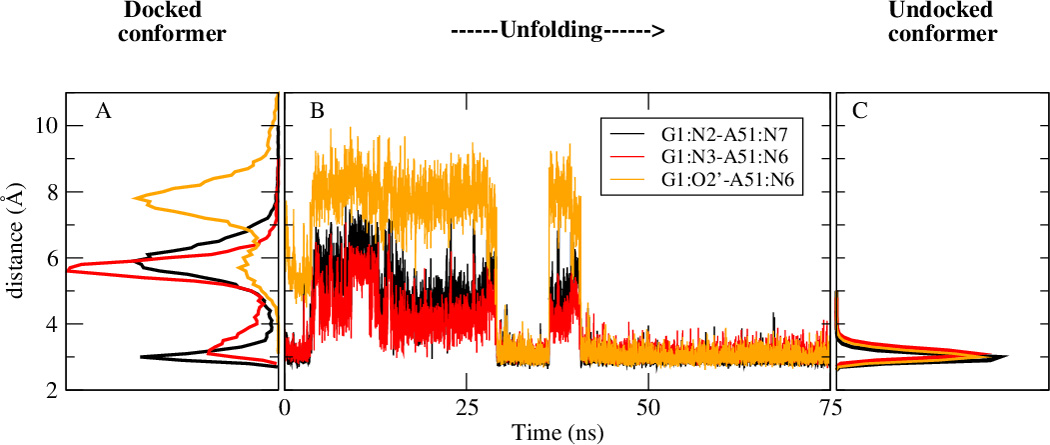
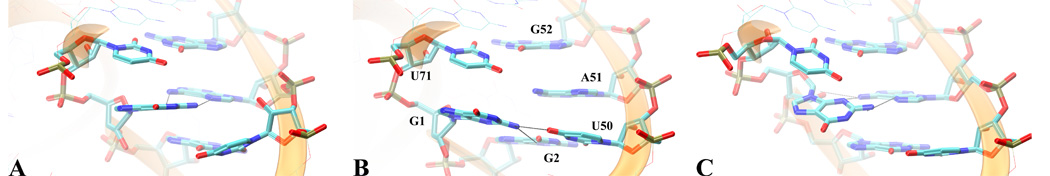
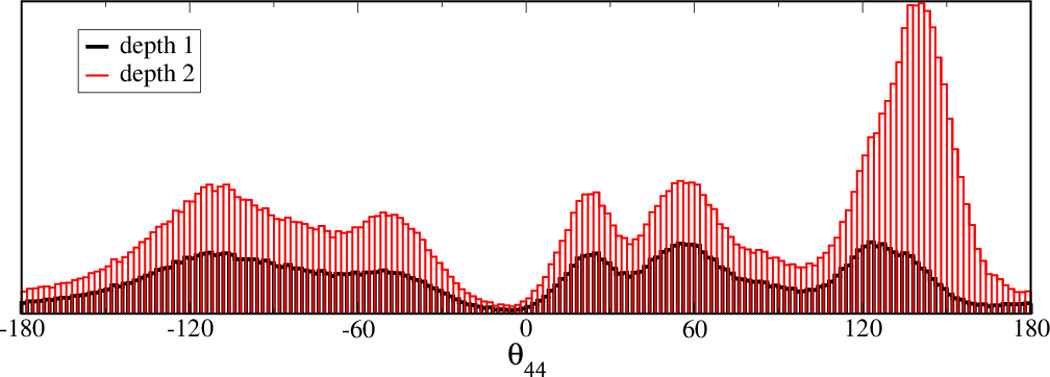
L1 ligase (L1L) molecular switch is an in vitro optimized synthetic allosteric ribozyme that catalyzes the regioselective formation of a 5'-to-3' phosphodiester bond, a reaction for which there is no known naturally occurring RNA catalyst. L1L serves as a proof of principle that RNA can catalyze a critical reaction for prebiotic RNA self-replication according to the RNA world hypothesis. L1L crystal structure captures two distinct conformations that differ by a reorientation of one of the stems by around 80Å and are presumed to correspond to the active and inactive state, respectively. It is of great interest to understand the nature of these two states in solution and the pathway for their interconversion. In this study, we use explicit solvent molecular simulation together with a novel enhanced sampling method that utilizes concepts from network theory to map out the conformational transition between active and inactive states of L1L. We find that the overall switching mechanism can be described as a three-state/two-step process. The first step involves a large-amplitude swing that reorients stem C. The second step involves the allosteric activation of the catalytic site through distant contacts with stem C. Using a conformational space network representation of the L1L switch transition, it is shown that the connection between the three states follows different topographical patterns: the stem C swing step passes through a narrow region of the conformational space network, whereas the allosteric activation step covers a much wider region and a more diverse set of pathways through the network.
Copyright © 2012 Elsevier Ltd. All rights reserved.
Figures
Similar articles
-
Identification of dynamical hinge points of the L1 ligase molecular switch.RNA. 2010 Apr;16(4):769-80. doi: 10.1261/rna.1897810. Epub 2010 Feb 18. RNA. 2010. PMID: 20167653 Free PMC article.
-
The structural basis of ribozyme-catalyzed RNA assembly.Science. 2007 Mar 16;315(5818):1549-53. doi: 10.1126/science.1136231. Science. 2007. PMID: 17363667
-
Redesign of an artificial ligase ribozyme based on the analysis of its structural elements.RNA Biol. 2005 Oct-Dec;2(4):137-42. doi: 10.4161/rna.2.4.2302. Epub 2005 Nov 9. RNA Biol. 2005. PMID: 17114929
-
Crystallographic structures of the hammerhead ribozyme: relationship to ribozyme folding and catalysis.Annu Rev Biophys Biomol Struct. 1998;27:475-502. doi: 10.1146/annurev.biophys.27.1.475. Annu Rev Biophys Biomol Struct. 1998. PMID: 9646875 Review.
-
Catalytic strategies of self-cleaving ribozymes.Acc Chem Res. 2008 Aug;41(8):1027-35. doi: 10.1021/ar800050c. Epub 2008 Jul 25. Acc Chem Res. 2008. PMID: 18652494 Review.
Cited by
-
Multiscale methods for computational RNA enzymology.Methods Enzymol. 2015;553:335-74. doi: 10.1016/bs.mie.2014.10.064. Epub 2015 Jan 22. Methods Enzymol. 2015. PMID: 25726472 Free PMC article.
-
Native states of fast-folding proteins are kinetic traps.J Am Chem Soc. 2013 Mar 27;135(12):4729-34. doi: 10.1021/ja311077u. Epub 2013 Mar 15. J Am Chem Soc. 2013. PMID: 23458553 Free PMC article.
-
Prebiotic Factors Influencing the Activity of a Ligase Ribozyme.Life (Basel). 2017 Apr 6;7(2):17. doi: 10.3390/life7020017. Life (Basel). 2017. PMID: 28383486 Free PMC article.
-
RNA Structural Dynamics As Captured by Molecular Simulations: A Comprehensive Overview.Chem Rev. 2018 Apr 25;118(8):4177-4338. doi: 10.1021/acs.chemrev.7b00427. Epub 2018 Jan 3. Chem Rev. 2018. PMID: 29297679 Free PMC article. Review.
-
Efficient in silico exploration of RNA interhelical conformations using Euler angles and WExplore.Nucleic Acids Res. 2014 Oct 29;42(19):12126-37. doi: 10.1093/nar/gku799. Epub 2014 Oct 7. Nucleic Acids Res. 2014. PMID: 25294827 Free PMC article.
References
-
- Allen M, Tildesley D. Computer Simulation of Liquids. Oxford: Oxford University Press; 1987.
-
- Bailor MH, Sun X, Al-Hashimi HM. Topology links RNA secondary structure with global conformation, dynamics, and adaptation. Science. 2010 Jan;327(5962):202–206. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
