

Lung cancers with acquired resistance to EGFR inhibitors occasionally harbor BRAF gene mutations but lack mutations in KRAS, NRAS, or MEK1
- PMID: 22773810
- PMCID: PMC3411967
- DOI: 10.1073/pnas.1203530109
Lung cancers with acquired resistance to EGFR inhibitors occasionally harbor BRAF gene mutations but lack mutations in KRAS, NRAS, or MEK1
Abstract
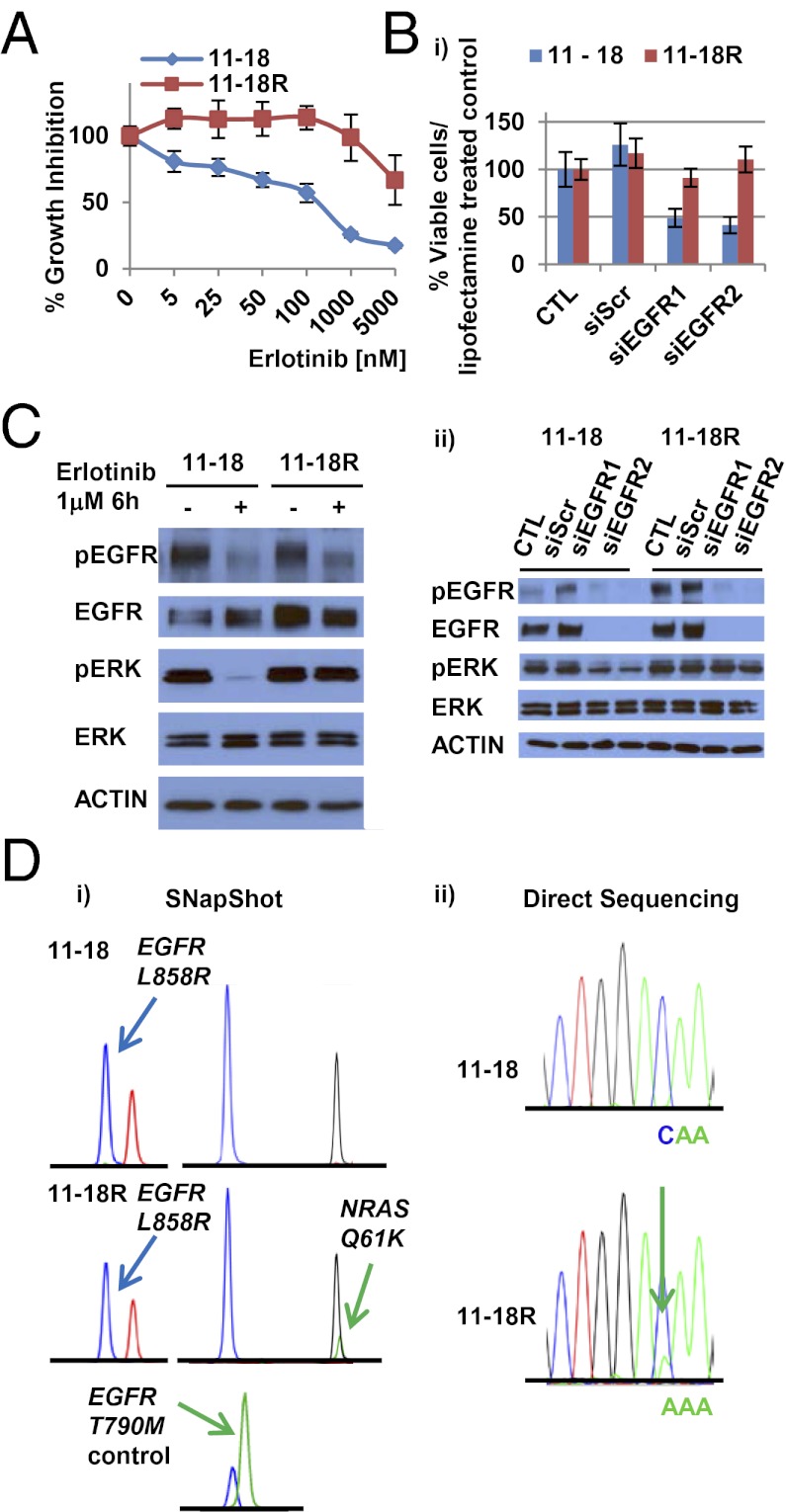
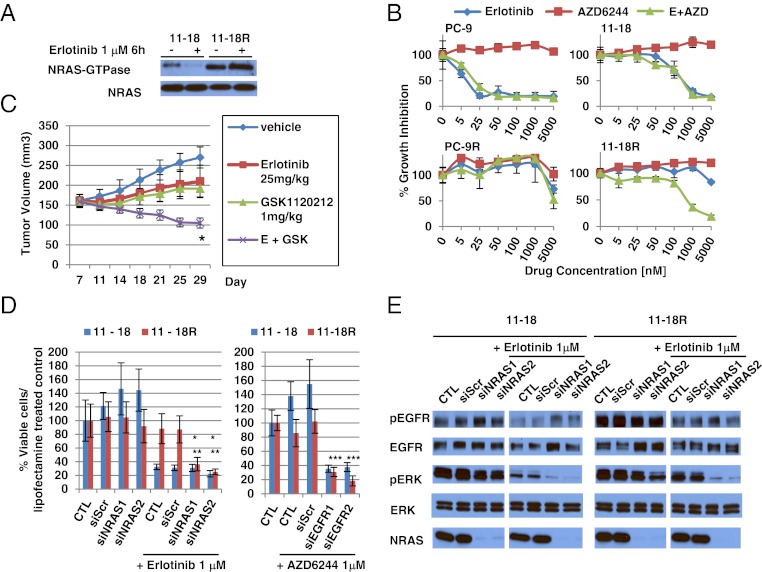
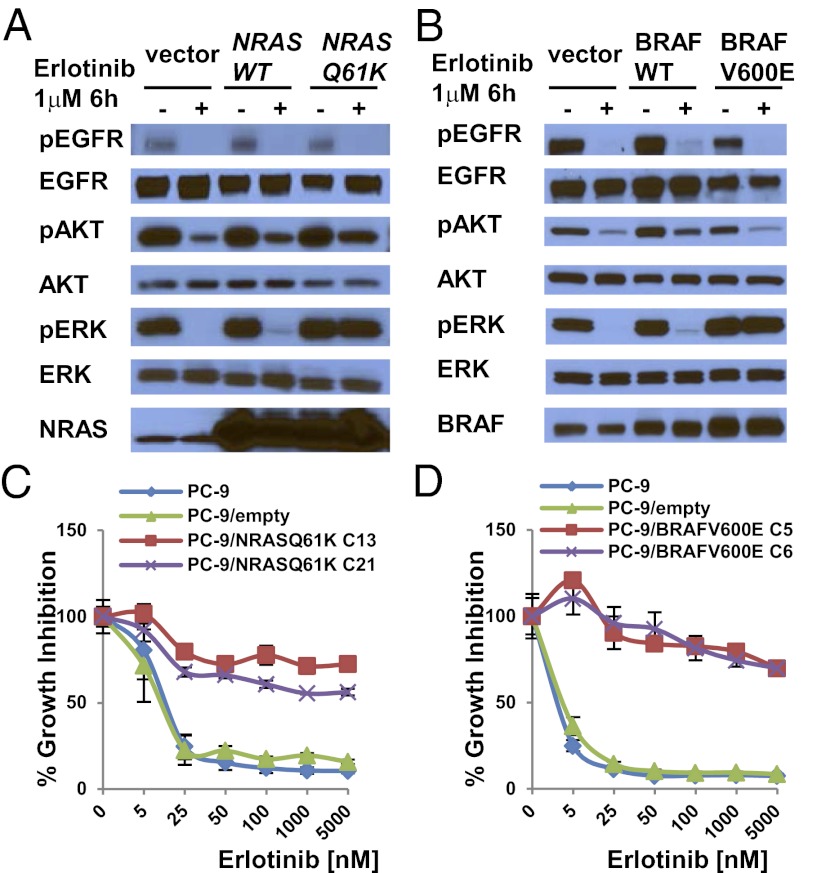
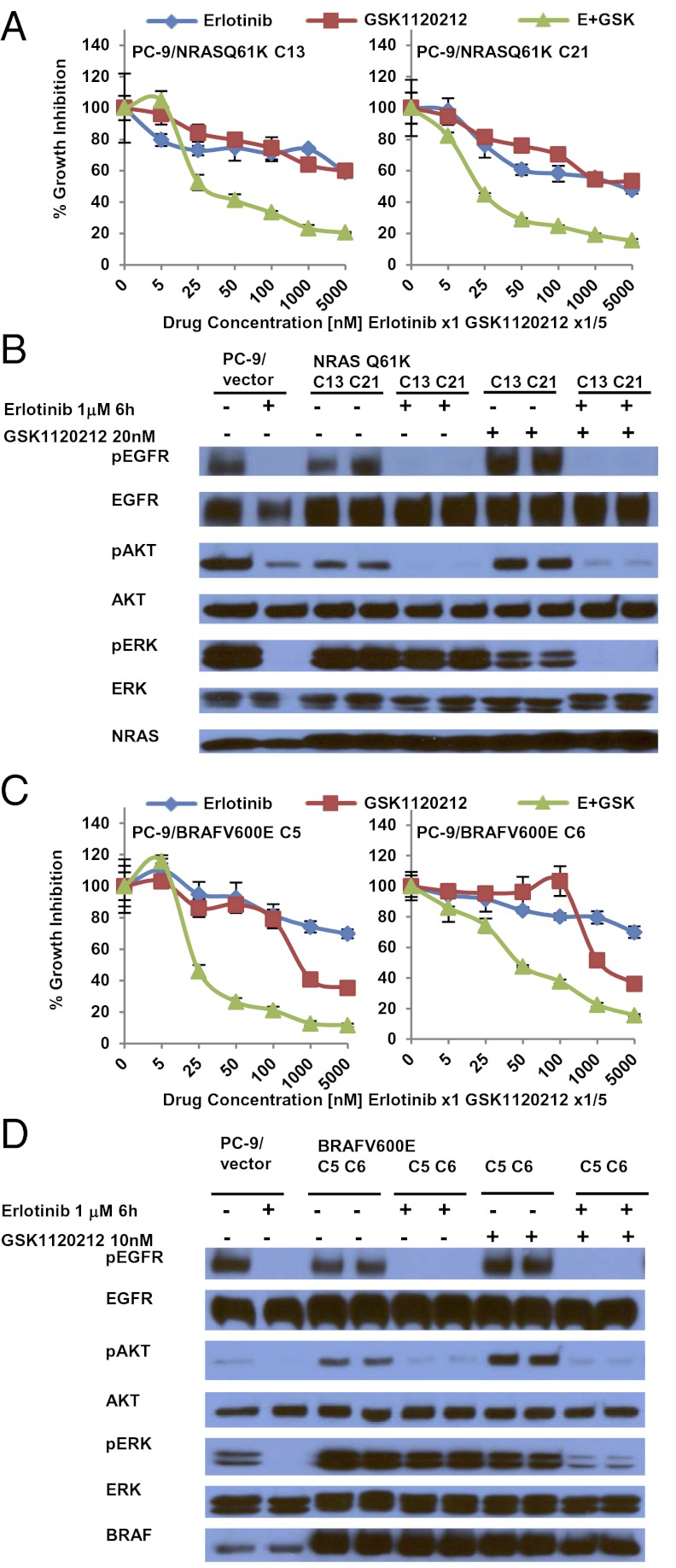
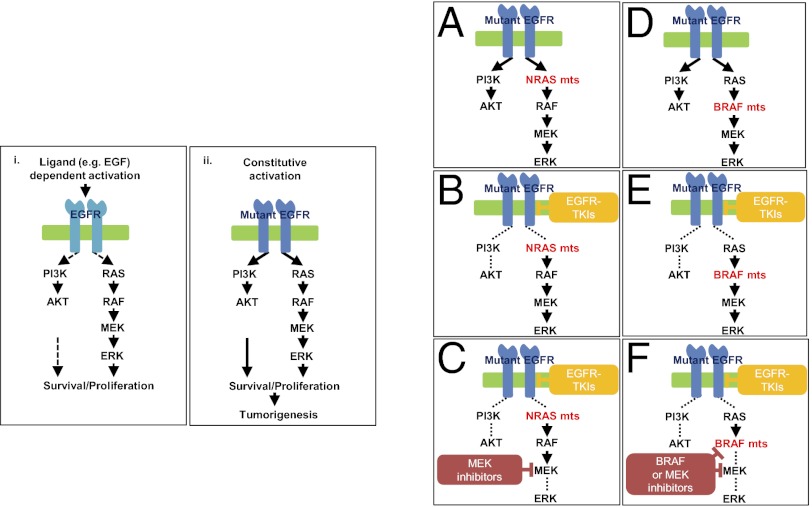
Acquired resistance to EGF receptor (EGFR) tyrosine kinase inhibitors (TKIs) is inevitable in metastatic EGFR-mutant lung cancers. Here, we modeled disease progression using EGFR-mutant human tumor cell lines. Although five of six models displayed alterations already found in humans, one harbored an unexpected secondary NRAS Q61K mutation; resistant cells were sensitive to concurrent EGFR and MEK inhibition but to neither alone. Prompted by this finding and because RAS/RAF/MEK mutations are known mediators of acquired resistance in other solid tumors (colon cancers, gastrointestinal stromal tumors, and melanomas) responsive to targeted therapies, we analyzed the frequency of secondary KRAS/NRAS/BRAF/MEK1 gene mutations in the largest collection to date of lung cancers with acquired resistance to EGFR TKIs. No recurrent NRAS, KRAS, or MEK1 mutations were found in 212, 195, or 146 patient samples, respectively, but 2 of 195 (1%) were found to have mutations in BRAF (G469A and V600E). Ectopic expression of mutant NRAS or BRAF in drug-sensitive EGFR-mutant cells conferred resistance to EGFR TKIs that was overcome by addition of a MEK inhibitor. Collectively, these positive and negative results provide deeper insight into mechanisms of acquired resistance to EGFR TKIs in lung cancer and inform ongoing clinical trials designed to overcome resistance. In the context of emerging knowledge about mechanisms of acquired resistance to targeted therapies in various cancers, our data highlight the notion that, even though solid tumors share common signaling cascades, mediators of acquired resistance must be elucidated for each disease separately in the context of treatment.
Conflict of interest statement
Conflict of interest statement: L.V.S. received consulting fees from Clovis Oncology, GlaxoSmithKline, and Celgene Corporation. J.C.-H.Y. received consulting fees from Boehringer Ingelheim. V.A.M. is an employee at Foundation Medicine and has equity value in the company. M.G.K. has received consulting fees from Boehringer Ingelheim and research funding for other projects from Pfizer and Boehringer. J.A.E. has received consulting fees and has stock option ownership in Agios Pharmaceuticals and has received research funding for other projects from Novartis, GlaxoSmithKline, and AstraZeneca. D.D.-S. received consulting fees from Bio-Reference Laboratories. W.P. has received consulting fees from MolecularMD, AstraZeneca, Bristol-Myers Squibb, Symphony Evolution, and Clovis Oncology and research funding for other projects from Enzon, Xcovery, AstraZeneca, and Symphogen. W.P. and V.A.M. are part of a patent regarding EGFR T790M mutation testing that was licensed by Memorial Sloan-Kettering Cancer Center to MolecularMD.
Figures
References
-
- Lynch TJ, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. - PubMed
-
- Paez JG, et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. - PubMed
-
- Shigematsu H, et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst. 2005;97:339–346. - PubMed
-
- Maemondo M, et al. North-East Japan Study Group Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362:2380–2388. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
