

Macrophage migration inhibitory factor deficiency protects pancreatic islets from cytokine-induced apoptosis in vitro
- PMID: 22774990
- PMCID: PMC3406375
- DOI: 10.1111/j.1365-2249.2012.04607.x
Macrophage migration inhibitory factor deficiency protects pancreatic islets from cytokine-induced apoptosis in vitro
Abstract
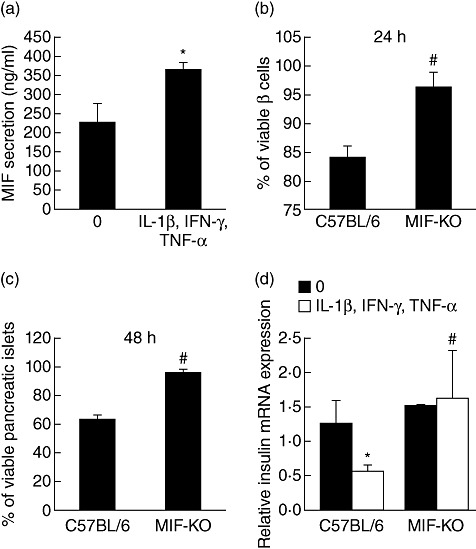
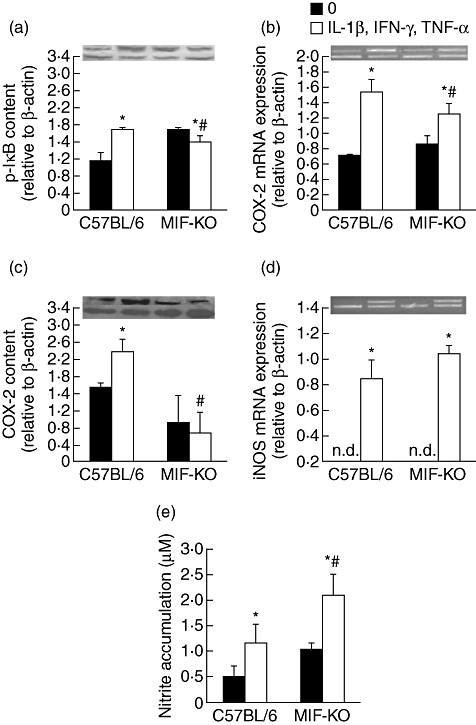
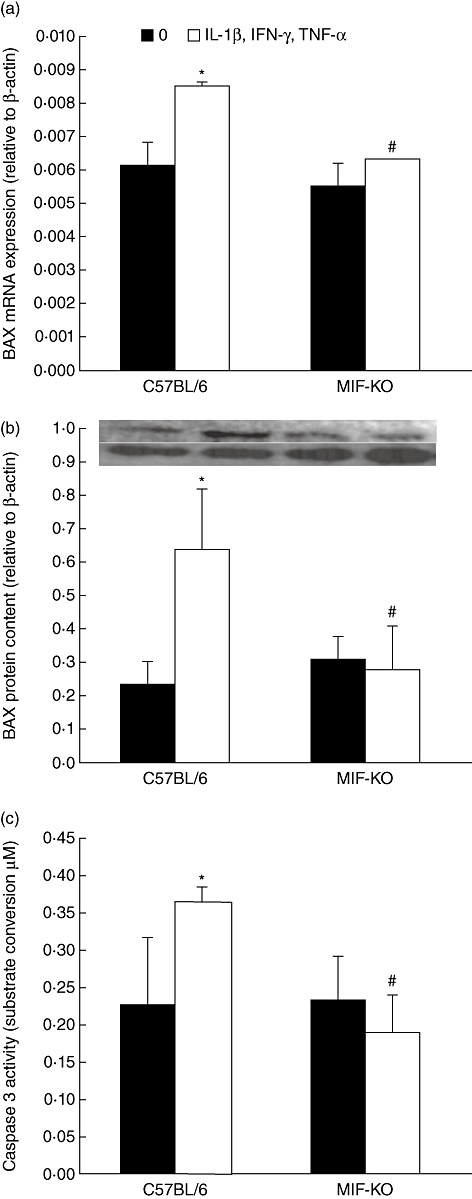
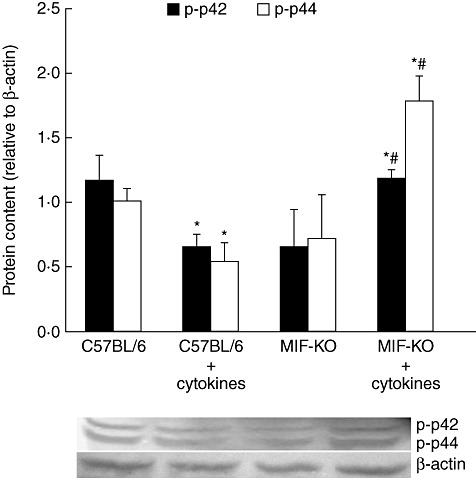
During pathogenesis of diabetes, pancreatic islets are exposed to high levels of cytokines and other inflammatory mediators that induce deterioration of insulin-producing beta cells. Macrophage migration inhibitory factor (MIF) plays a key role in the onset and development of several immunoinflammatory diseases and also controls apoptotic cell death. Because the occurrence of apoptosis plays a pathogenetic role in beta cell death during type 1 diabetes development and MIF is expressed in beta cells, we explored the influence of MIF deficiency on cytokine-induced apoptosis in pancreatic islets. The results indicated clearly that elevated MIF secretion preceded C57BL/6 pancreatic islets death induced by interferon (IFN)-γ + tumour necrosis factor (TNF)-α + interleukin (IL)-1β. Consequently, MIF-deficient [MIF-knock-out (KO)] pancreatic islets or islet cells showed significant resistance to cytokine-induced death than those isolated from C57BL/6 mice. Furthermore, upon exposure to cytokines pancreatic islets from MIF-KO mice maintained normal insulin expression and produced less cyclooxygenase-2 (COX-2) than those from wild-type C57BL6 mice. The final outcome of cytokine-induced islet apoptosis in islets from wild-type mice was the activation of mitochondrial membrane pore-forming protein Bcl-2-associated X protein and effector caspase 3. In contrast, these apoptotic mediators remained at normal levels in islets from MIF-KO mice suggesting that MIF absence prevented initiation of the mitochondrial apoptotic pathway. Additionally, the protection from apoptosis was also mediated by up-regulation of prosurvival kinase extracellular-regulated kinase 1/2 in MIF-KO islets. These data indicate that MIF is involved in the propagation of pancreatic islets apoptosis probably via nuclear factor-κB and mitochondria-related proteins.
© 2012 The Authors. Clinical and Experimental Immunology © 2012 British Society for Immunology.
Figures
References
-
- Beyan H, Buckley LR, Yousaf N, Londei M, Leslie RD. A role for innate immunity in type 1 diabetes? Diabetes Metab Res Rev. 2003;19:89–100. - PubMed
-
- Kaminitz A, Stein J, Yaniv I, Askenasy N. The vicious cycle of apoptotic β-cell death in type 1 diabetes. Immunol Cell Biol. 2007;85:582–9. - PubMed
-
- Pirot P, Cardozo AK, Eizirik DL. Mediators and mechanisms of pancreatic beta-cell death in type 1 diabetes. Arq Bras Endocrinol Metabol. 2008;52:156–65. - PubMed
-
- Davi G, Falco A, Patrono C. Lipid peroxidation in diabetes mellitus. Antioxid Redox Signal. 2005;7:256–68. - PubMed
-
- Eizirik DL, Colli ML, Ortis F. The role of inflammation in insulitis and beta-cell loss in type 1 diabetes. Nat Rev Endocrinol. 2009;5:219–26. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
