

Assessment of the structural and functional impact of in-frame mutations of the DMD gene, using the tools included in the eDystrophin online database
- PMID: 22776072
- PMCID: PMC3748829
- DOI: 10.1186/1750-1172-7-45
Assessment of the structural and functional impact of in-frame mutations of the DMD gene, using the tools included in the eDystrophin online database
Abstract
Background: Dystrophin is a large essential protein of skeletal and heart muscle. It is a filamentous scaffolding protein with numerous binding domains. Mutations in the DMD gene, which encodes dystrophin, mostly result in the deletion of one or several exons and cause Duchenne (DMD) and Becker (BMD) muscular dystrophies. The most common DMD mutations are frameshift mutations resulting in an absence of dystrophin from tissues. In-frame DMD mutations are less frequent and result in a protein with partial wild-type dystrophin function. The aim of this study was to highlight structural and functional modifications of dystrophin caused by in-frame mutations.
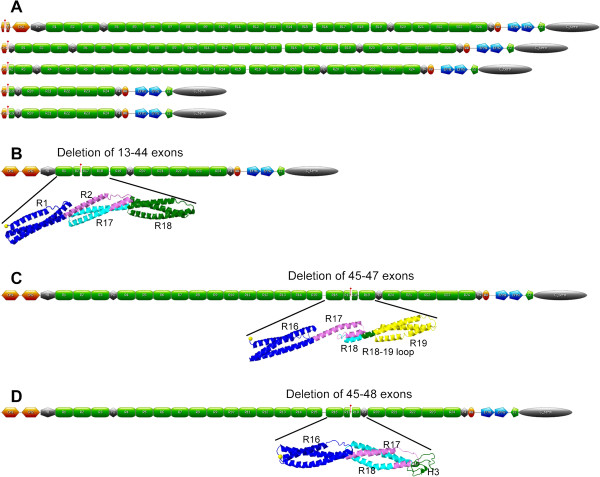
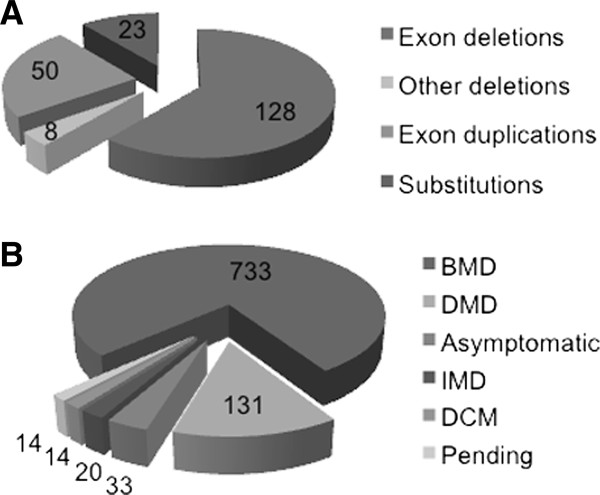
Methods and results: We developed a dedicated database for dystrophin, the eDystrophin database. It contains 209 different non frame-shifting mutations found in 945 patients from a French cohort and previous studies. Bioinformatics tools provide models of the three-dimensional structure of the protein at deletion sites, making it possible to determine whether the mutated protein retains the typical filamentous structure of dystrophin. An analysis of the structure of mutated dystrophin molecules showed that hybrid repeats were reconstituted at the deletion site in some cases. These hybrid repeats harbored the typical triple coiled-coil structure of native repeats, which may be correlated with better function in muscle cells.
Conclusion: This new database focuses on the dystrophin protein and its modification due to in-frame deletions in BMD patients. The observation of hybrid repeat reconstitution in some cases provides insight into phenotype-genotype correlations in dystrophin diseases and possible strategies for gene therapy. The eDystrophin database is freely available: http://edystrophin.genouest.org/.
Figures




References
-
- Mendelian Inheritance in Man, [http://www.ncbi.nlm.gov/omim]
-
- Segalat L. Dystrophin and functionally related proteins in the nematode Caenorhabditis elegans. Neuromuscul Disord. 2002;12(Suppl 1):S105–S109. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
