

RAD51 mutants cause replication defects and chromosomal instability
- PMID: 22778135
- PMCID: PMC3430192
- DOI: 10.1128/MCB.00406-12
RAD51 mutants cause replication defects and chromosomal instability
Abstract
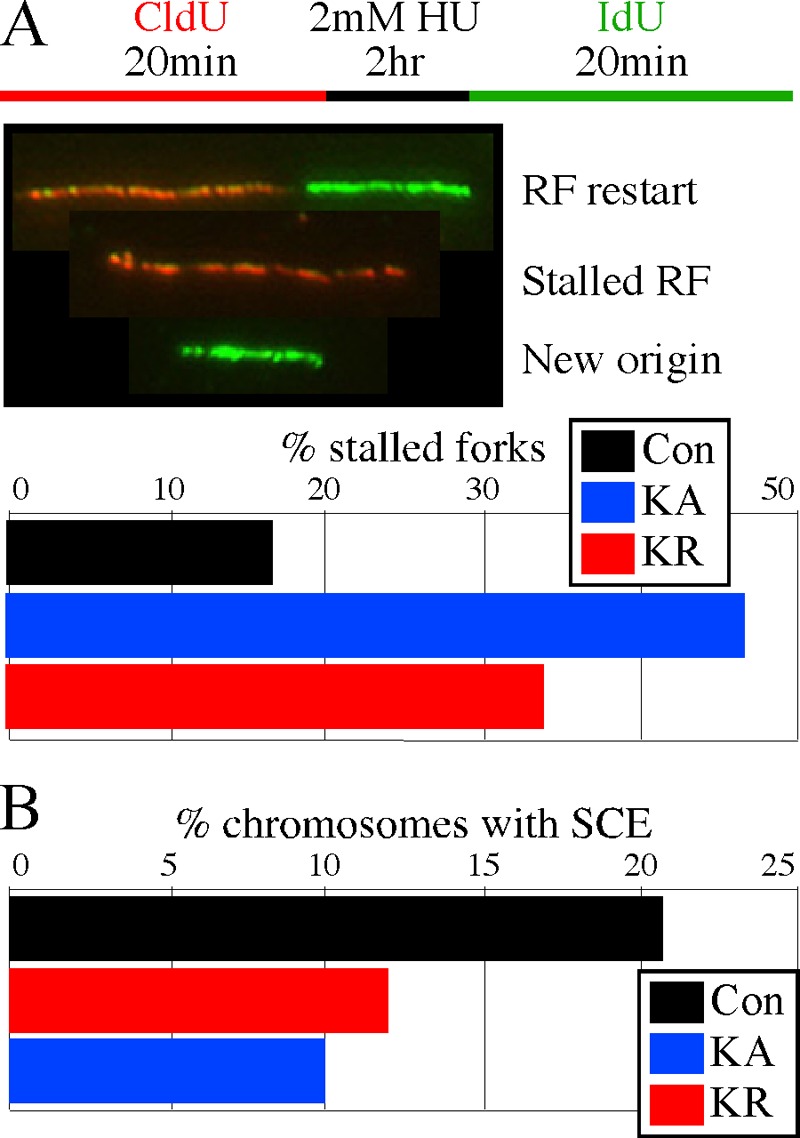
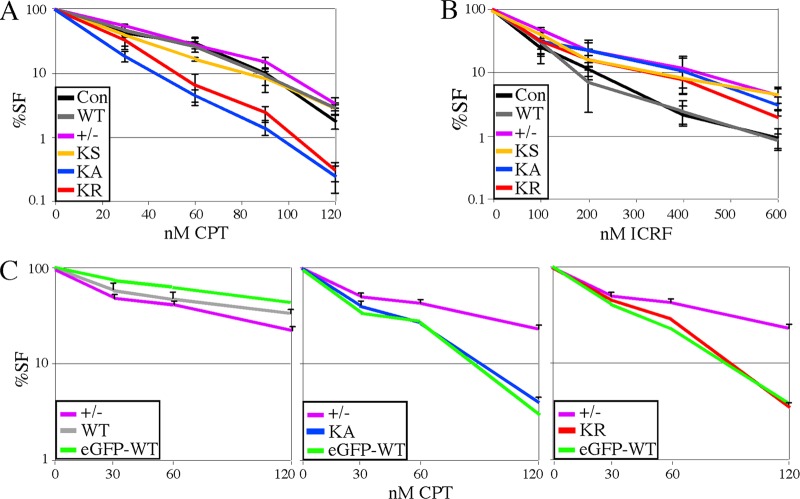
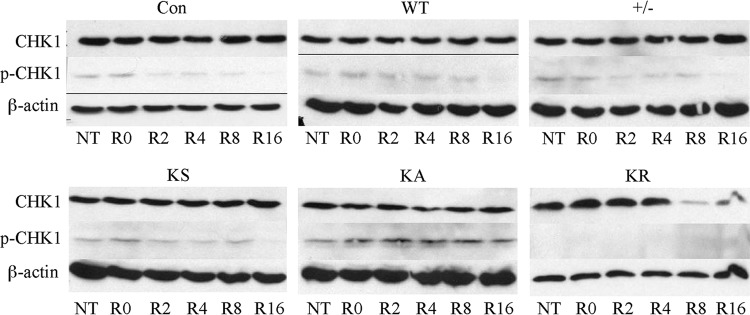
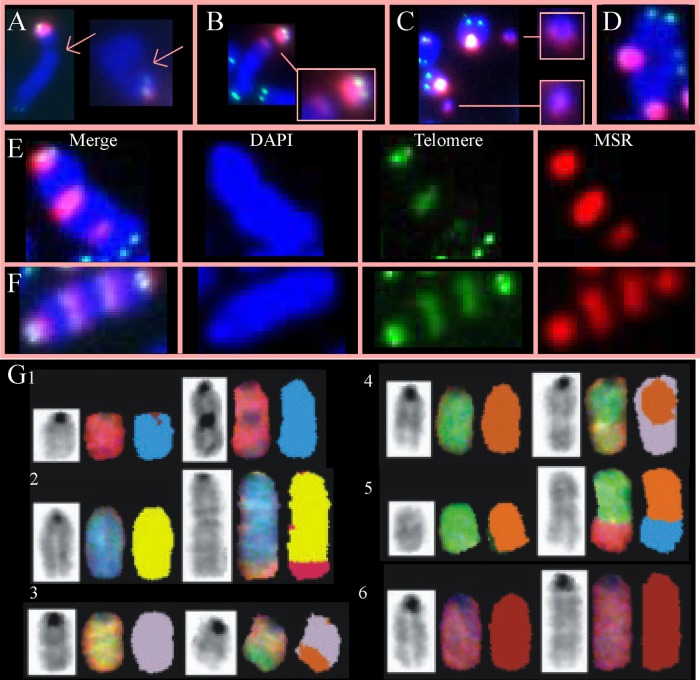
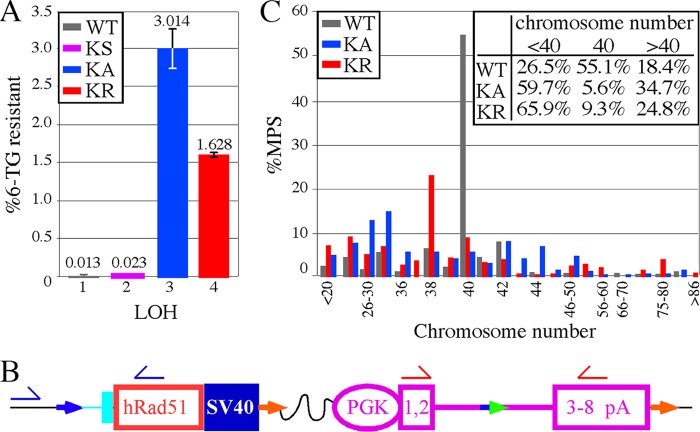
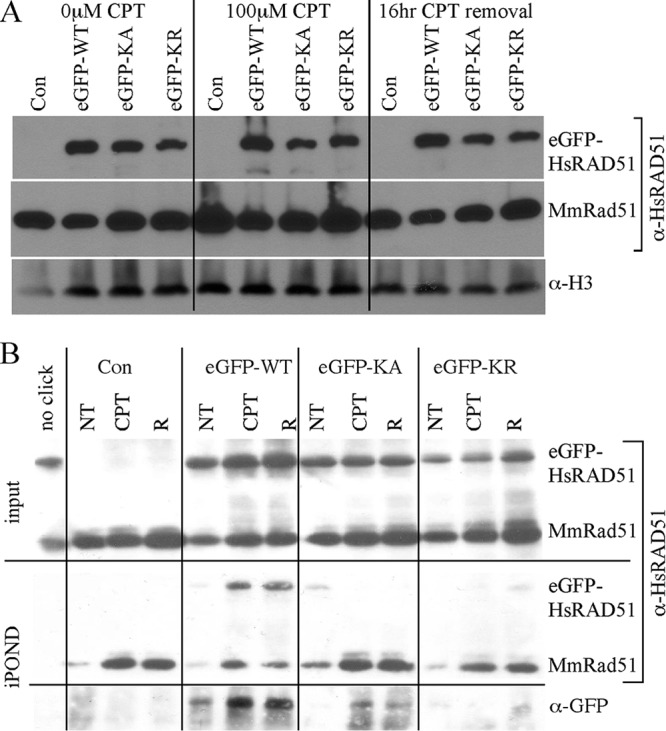
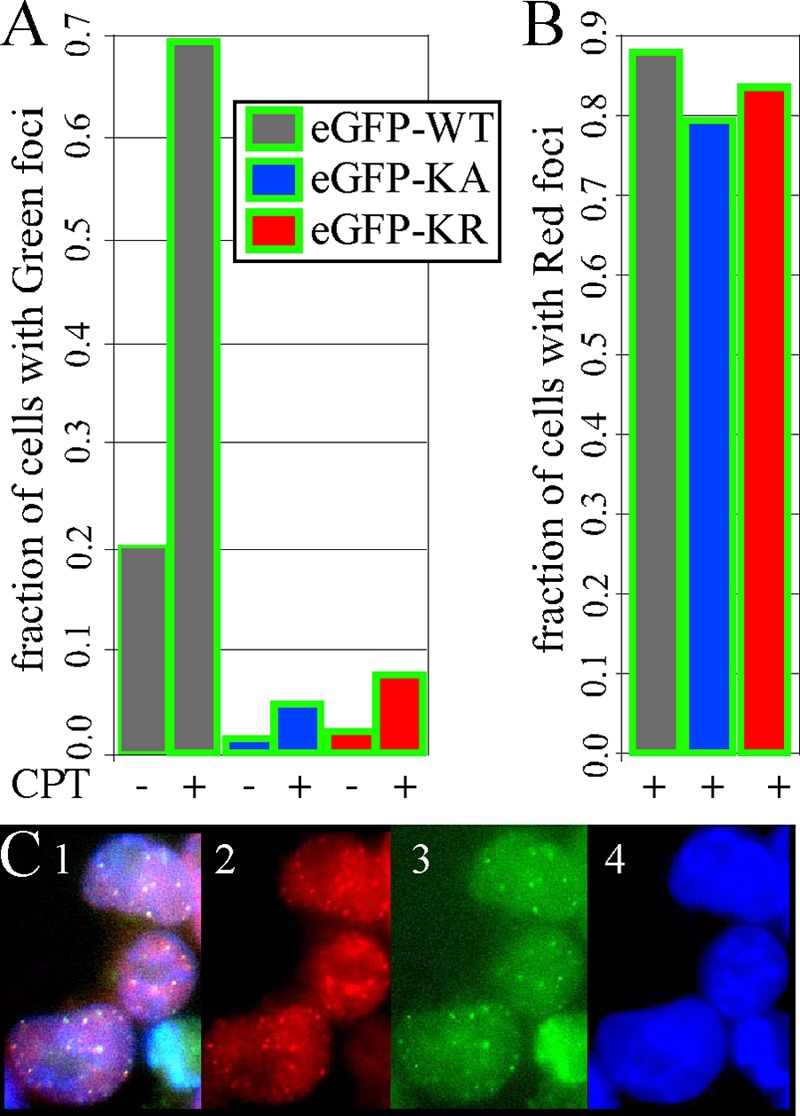
RAD51 is important for restarting stalled replication forks and for repairing DNA double-strand breaks (DSBs) through a pathway called homology-directed repair (HDR). However, analysis of the consequences of specific RAD51 mutants has been difficult since they are toxic. Here we report on the dominant effects of two human RAD51 mutants defective for ATP binding (K133A) or ATP hydrolysis (K133R) expressed in mouse embryonic stem (ES) cells that also expressed normal mouse RAD51 from the other chromosome. These cells were defective for restarting stalled replication forks and repairing breaks. They were also hypersensitive to camptothecin, a genotoxin that generates breaks specifically at the replication fork. In addition, these cells exhibited a wide range of structural chromosomal changes that included multiple breakpoints within the same chromosome. Thus, ATP binding and hydrolysis are essential for chromosomal maintenance. Fusion of RAD51 to a fluorescent tag (enhanced green fluorescent protein [eGFP]) allowed visualization of these proteins at sites of replication and repair. We found very low levels of mutant protein present at these sites compared to normal protein, suggesting that low levels of mutant protein were sufficient for disruption of RAD51 activity and generation of chromosomal rearrangements.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials