

Transcriptome and methylome interactions in rice hybrids
- PMID: 22778444
- PMCID: PMC3409791
- DOI: 10.1073/pnas.1209297109
Transcriptome and methylome interactions in rice hybrids
Abstract
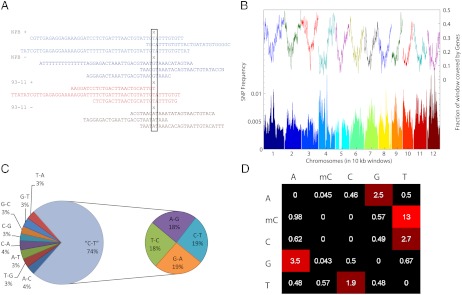
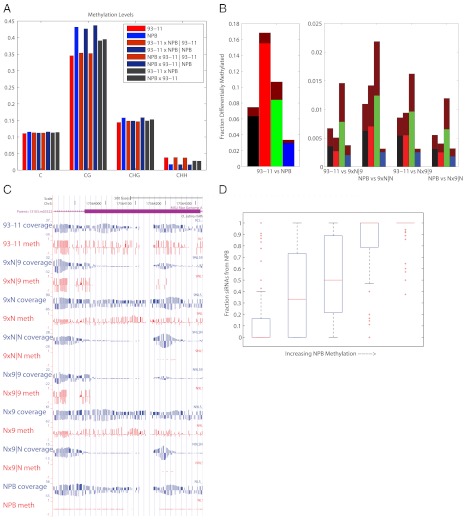
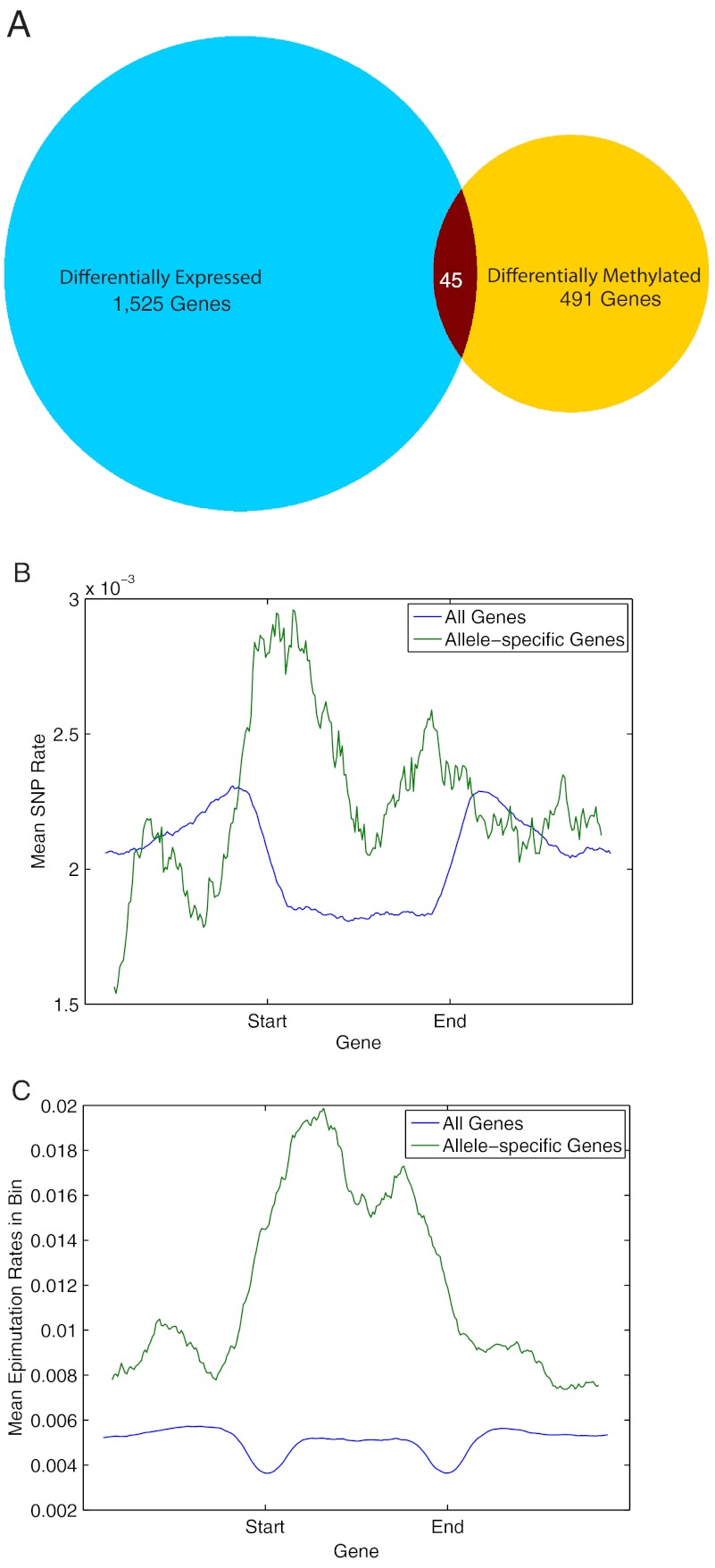
DNA methylation is a heritable epigenetic mark that controls gene expression, is responsive to environmental stresses, and, in plants, may also play a role in heterosis. To determine the degree to which DNA methylation is inherited in rice, and how it both influences and is affected by transcription, we performed genome-wide measurements of these patterns through an integrative analysis of bisulfite-sequencing, RNA-sequencing, and siRNA-sequencing data in two inbred parents of the Nipponbare (NPB) and indica (93-11) varieties of rice and their hybrid offspring. We show that SNPs occur at a rate of about 1/253 bp between the two parents and that these are faithfully transmitted into the hybrids. We use the presence of these SNPs to reconstruct the two chromosomes in the hybrids according to their parental origin. We found that, unlike genetic inheritance, epigenetic heritability is quite variable. Cytosines were found to be differentially methylated (epimutated) at a rate of 7.48% (1/15 cytosines) between the NPB and 93-11 parental strains. We also observed that 0.79% of cytosines were epimutated between the parent and corresponding hybrid chromosome. We found that these epimutations are often clustered on the chromosomes, with clusters representing 20% of all epimutations between parental ecotypes, and 2-5% in F1 plants. Epimutation clusters are also strongly associated with regions where the production of siRNA differs between parents. Finally, we identified genes with both allele-specific expression patterns that were strongly inherited as well as those differentially expressed between hybrids and the corresponding parental chromosome. We conclude that much of the misinheritance of expression levels is likely caused by epimutations and trans effects.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Bird A. Perceptions of epigenetics. Nature. 2007;447:396–398. - PubMed
-
- Suzuki MM, Bird A. DNA methylation landscapes: Provocative insights from epigenomics. Nat Rev Genet. 2008;9:465–476. - PubMed
-
- Zhang X, et al. Genome-wide high-resolution mapping and functional analysis of DNA methylation in Arabidopsis. Cell. 2006;126:1189–1201. - PubMed
-
- Zilberman D, Gehring M, Tran RK, Ballinger T, Henikoff S. Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat Genet. 2007;39:61–69. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
