

Predicting new indications for approved drugs using a proteochemometric method
- PMID: 22780961
- PMCID: PMC3419493
- DOI: 10.1021/jm300576q
Predicting new indications for approved drugs using a proteochemometric method
Abstract
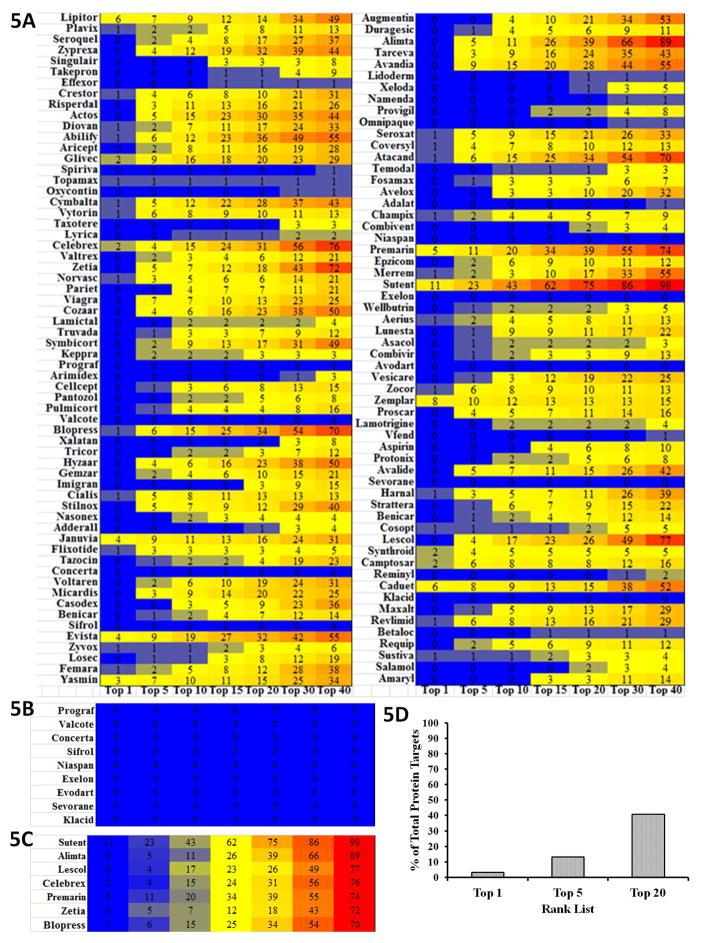
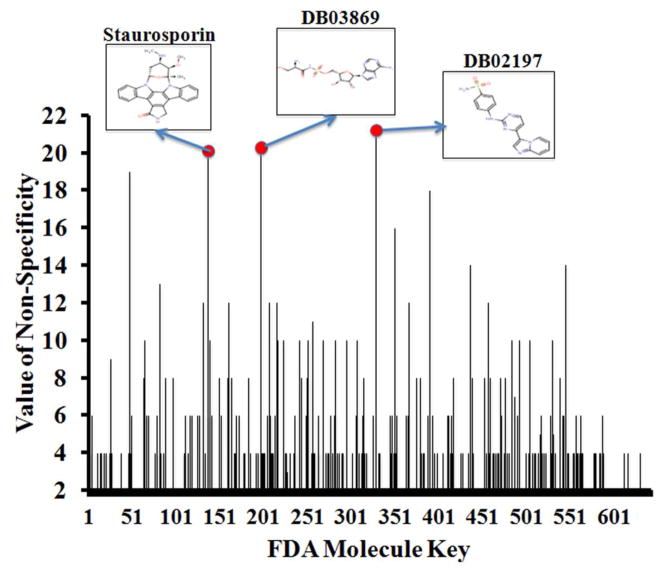
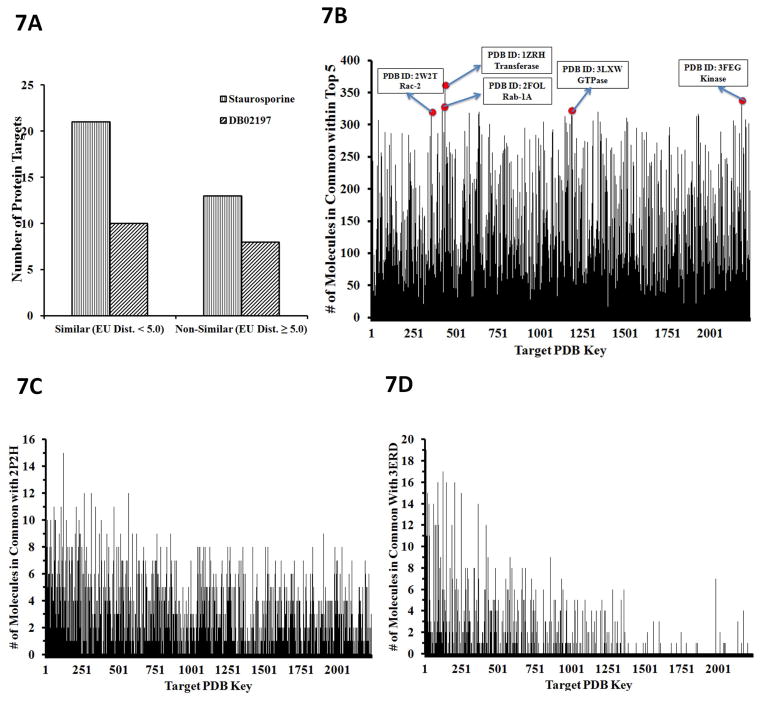
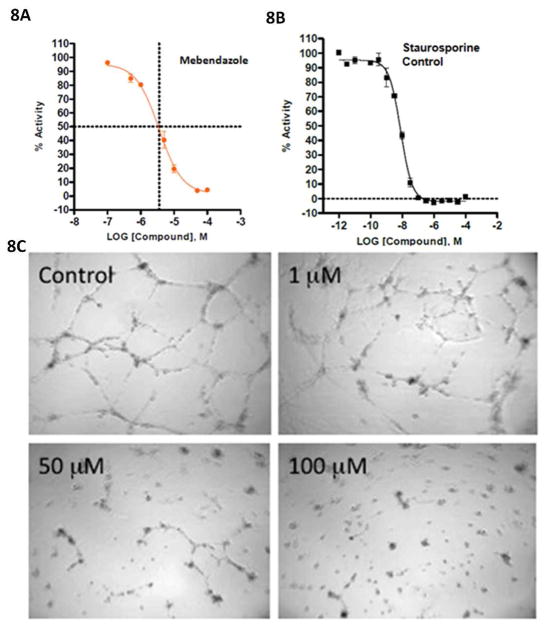
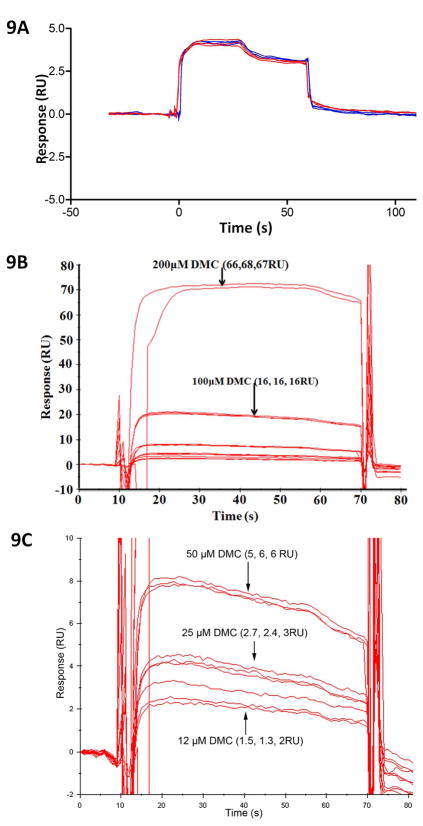
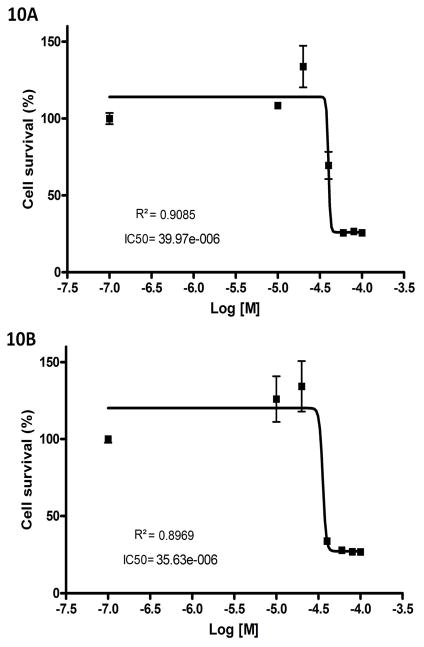
The most effective way to move from target identification to the clinic is to identify already approved drugs with the potential for activating or inhibiting unintended targets (repurposing or repositioning). This is usually achieved by high throughput chemical screening, transcriptome matching, or simple in silico ligand docking. We now describe a novel rapid computational proteochemometric method called "train, match, fit, streamline" (TMFS) to map new drug-target interaction space and predict new uses. The TMFS method combines shape, topology, and chemical signatures, including docking score and functional contact points of the ligand, to predict potential drug-target interactions with remarkable accuracy. Using the TMFS method, we performed extensive molecular fit computations on 3671 FDA approved drugs across 2335 human protein crystal structures. The TMFS method predicts drug-target associations with 91% accuracy for the majority of drugs. Over 58% of the known best ligands for each target were correctly predicted as top ranked, followed by 66%, 76%, 84%, and 91% for agents ranked in the top 10, 20, 30, and 40, respectively, out of all 3671 drugs. Drugs ranked in the top 1-40 that have not been experimentally validated for a particular target now become candidates for repositioning. Furthermore, we used the TMFS method to discover that mebendazole, an antiparasitic with recently discovered and unexpected anticancer properties, has the structural potential to inhibit VEGFR2. We confirmed experimentally that mebendazole inhibits VEGFR2 kinase activity and angiogenesis at doses comparable with its known effects on hookworm. TMFS also predicted, and was confirmed with surface plasmon resonance, that dimethyl celecoxib and the anti-inflammatory agent celecoxib can bind cadherin-11, an adhesion molecule important in rheumatoid arthritis and poor prognosis malignancies for which no targeted therapies exist. We anticipate that expanding our TMFS method to the >27 000 clinically active agents available worldwide across all targets will be most useful in the repositioning of existing drugs for new therapeutic targets.
Figures
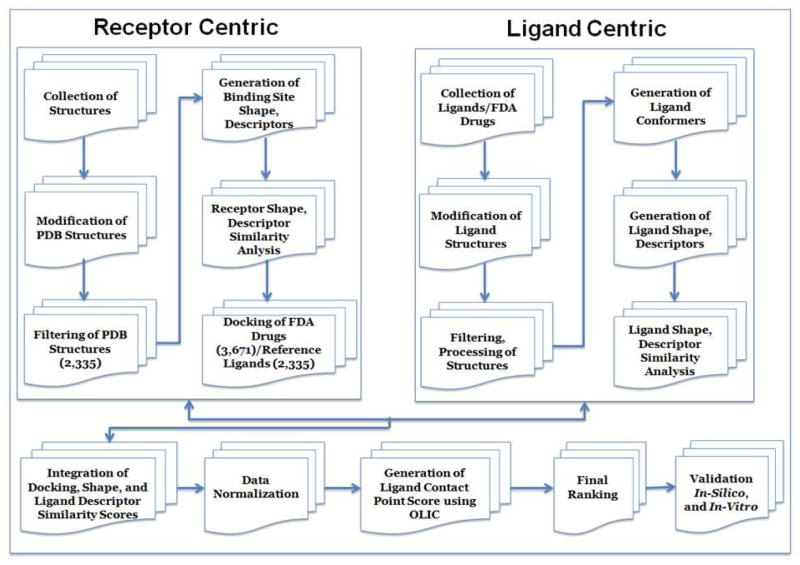
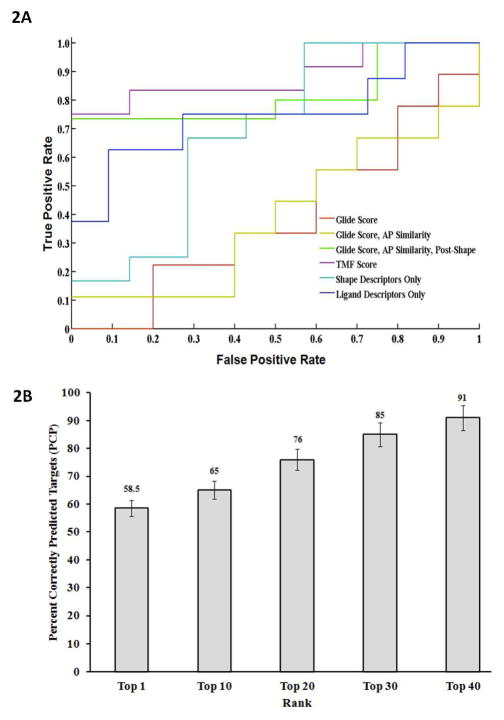
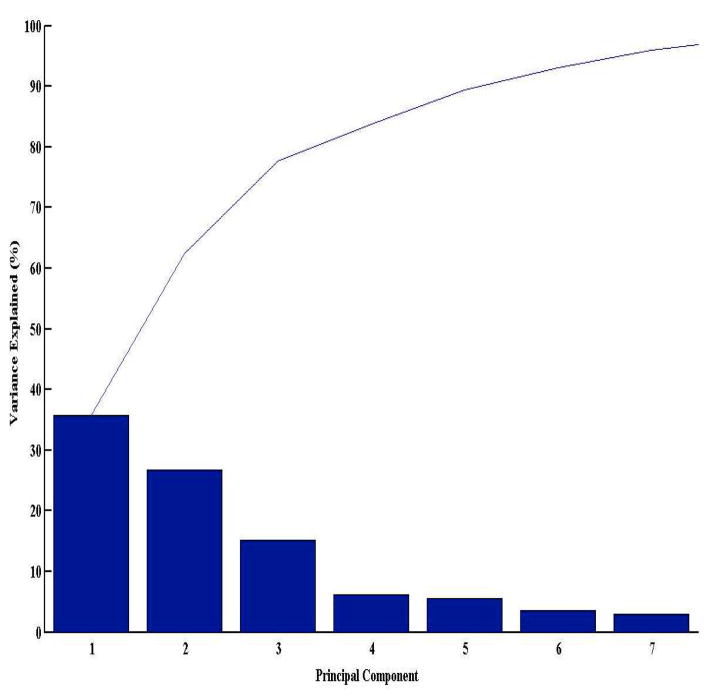
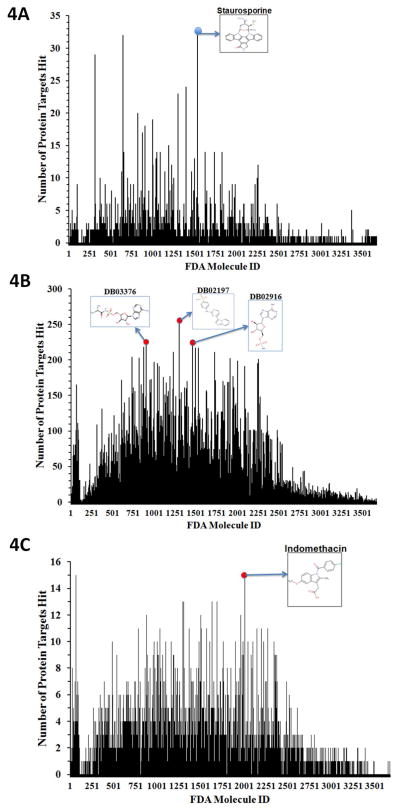
References
-
- Paul SM, Mytelka DS, Dunwiddie CT, Persinger CC, Munos BH, Lindborg SR, Schacht AL. How to improve R&D productivity: The pharmaceutical industry’s grand challenge. Nat Rev Drug Discovery. 2010;9:203–214. - PubMed
-
- Lawrence S. Drug output slows in 2006. Nat Biotechnol. 2007;25:1073.
-
- DiMasi JA, Hansen RW, Grabowski HG. The price of innovation: new estimates of drug development costs. J Health Econ. 2003;22(2):151–185. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Chemical Information
Medical
