

A novel LRSAM1 mutation is associated with autosomal dominant axonal Charcot-Marie-Tooth disease
- PMID: 22781092
- PMCID: PMC3548253
- DOI: 10.1038/ejhg.2012.146
A novel LRSAM1 mutation is associated with autosomal dominant axonal Charcot-Marie-Tooth disease
Abstract
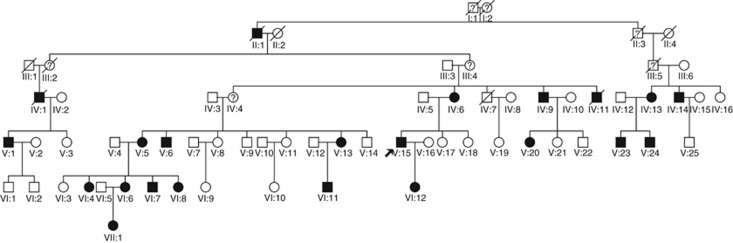
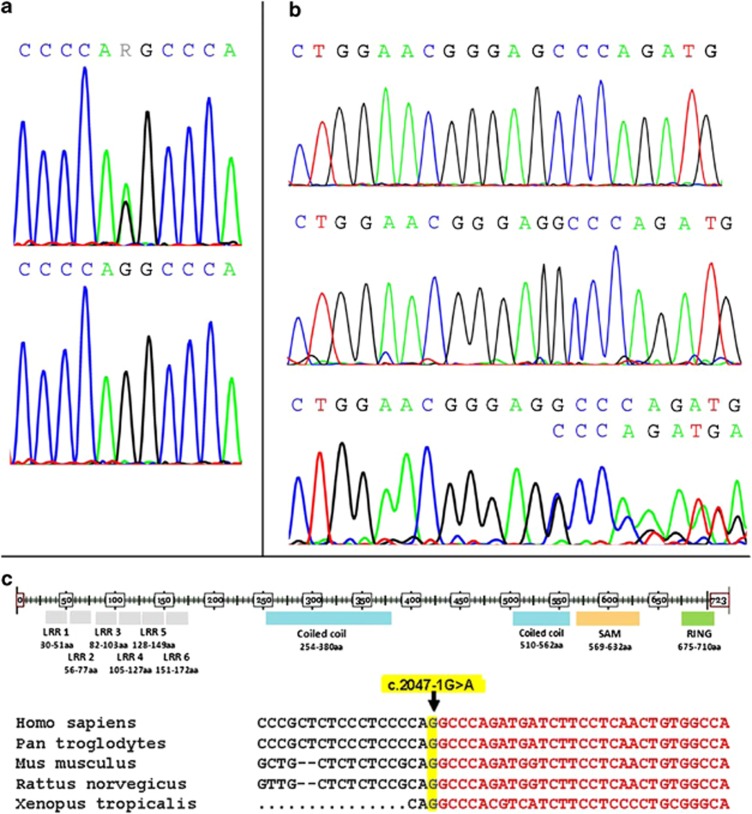
Charcot-Marie-Tooth (CMT) disease is the most common hereditary neuropathy resulting from mutations in >30 genes expressed in either the Schwann cells or the axon of peripheral nerves. The disease is classified into demyelinating (CMT1), axonal (CMT2) or intermediate (CMTI) based on electrophysiological and pathological findings. Our study focused on the identification of a novel disease mutation in a large Sardinian family with CMT2 of autosomal dominant (AD) inheritance. All available family members were clinically evaluated and samples were collected from consenting individuals. Initially, we excluded known CMT2 genes/loci in this family. We then conducted a genome-wide linkage analysis and mapped the gene to chromosome 9q33-q34. Refined linkage and haplotype analyses defined an 11.6-Mb candidate region with a maximum LOD score of 8.06. Following exclusion of several candidate genes from the region, we targeted the LRSAM1 (leucine-rich repeat and sterile alpha motif-containing 1) gene, very recently found to be associated with autosomal recessive CMT2 in one family. For a more efficient investigation of this large gene, already available proband RNA (cDNA) was initially analyzed. Targeted DNA analysis then confirmed a novel LRSAM1 splice-site (c.2047-1G>A) mutation, causing a frameshift that introduces a stop codon three amino acids further down the new reading frame (p.Ala683ProfsX3). This mutation is located in the C-terminal RING finger motif of the encoded protein and leads to premature truncation of the protein. In the course of our work, a second LRSAM1 mutation dominantly transmitted was identified by another group. Our data further confirms that LRSAM1 mutations are associated with CMT2 of AD inheritance.
Figures

References
-
- Barisic N, Claeys KG, Sirotkovic-Skerlev M, et al. Charcot-Marie-Tooth disease: a clinico-genetic confrontation. Ann Hum Genet. 2008;72:416–441. - PubMed
-
- Bird TD. Hereditary motor-sensory neuropathies. Charcot-Marie-Tooth syndrome. Neurol Clin. 1989;7:9–23. - PubMed
-
- Berger P, Young P, Suter U. Molecular cell biology of Charcot-Marie-Tooth disease. Neurogenetics. 2002;4:1–15. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
