

Novel findings in patients with primary hyperoxaluria type III and implications for advanced molecular testing strategies
- PMID: 22781098
- PMCID: PMC3548260
- DOI: 10.1038/ejhg.2012.139
Novel findings in patients with primary hyperoxaluria type III and implications for advanced molecular testing strategies
Abstract
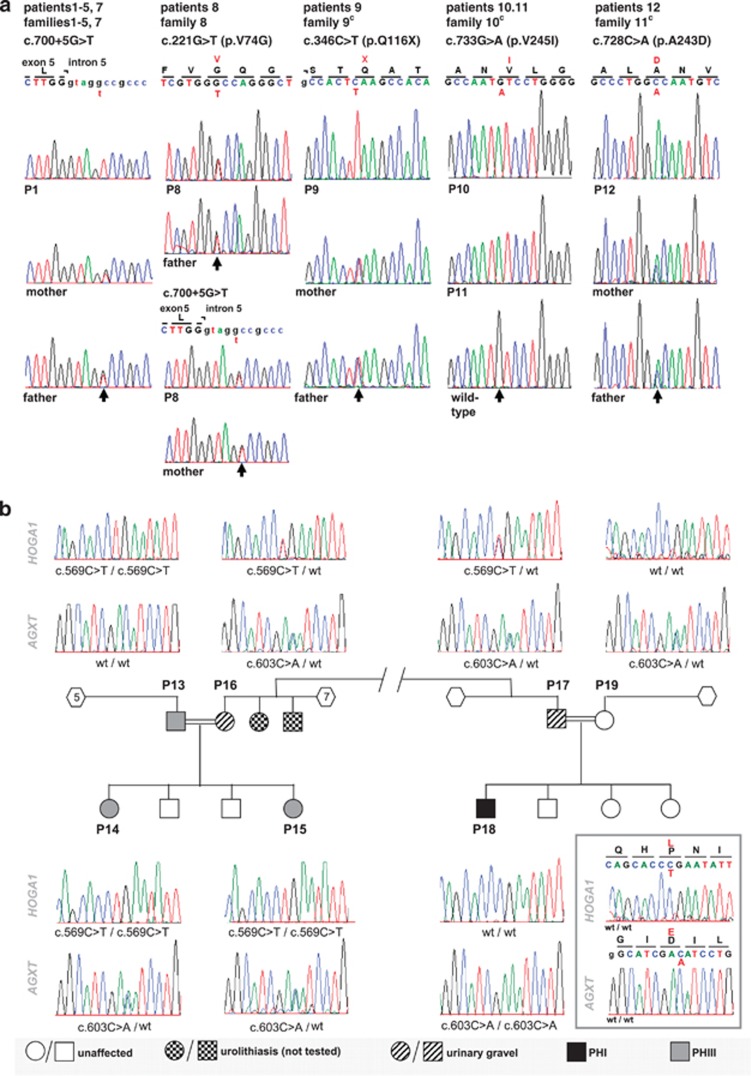
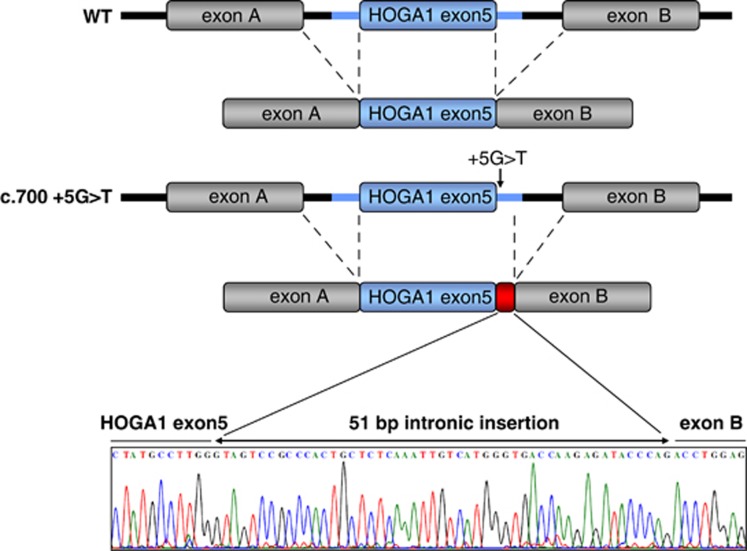
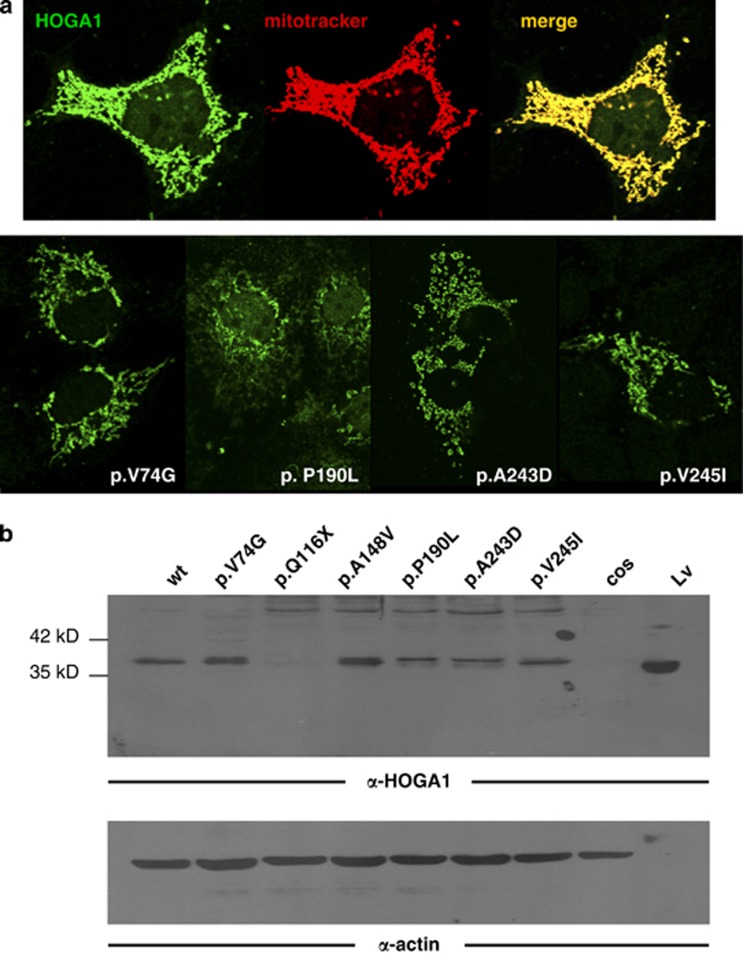
Identification of mutations in the HOGA1 gene as the cause of autosomal recessive primary hyperoxaluria (PH) type III has revitalized research in the field of PH and related stone disease. In contrast to the well-characterized entities of PH type I and type II, the pathophysiology and prevalence of type III is largely unknown. In this study, we analyzed a large cohort of subjects previously tested negative for type I/II by complete HOGA1 sequencing. Seven distinct mutations, among them four novel, were found in 15 patients. In patients of non-consanguineous European descent the previously reported c.700+5G>T splice-site mutation was predominant and represents a potential founder mutation, while in consanguineous families private homozygous mutations were identified throughout the gene. Furthermore, we identified a family where a homozygous mutation in HOGA1 (p.P190L) segregated in two siblings with an additional AGXT mutation (p.D201E). The two girls exhibiting triallelic inheritance presented a more severe phenotype than their only mildly affected p.P190L homozygous father. In silico analysis of five mutations reveals that HOGA1 deficiency is causing type III, yet reduced HOGA1 expression or aberrant subcellular protein targeting is unlikely to be the responsible pathomechanism. Our results strongly suggest HOGA1 as a major cause of PH, indicate a greater genetic heterogeneity of hyperoxaluria, and point to a favorable outcome of type III in the context of PH despite incomplete or absent biochemical remission. Multiallelic inheritance could have implications for genetic testing strategies and might represent an unrecognized mechanism for phenotype variability in PH.
Figures
References
-
- Archer HE, Dormer AE, Scowen EF, Watts RW. Primary hyperoxaluria. Lancet. 1957;273:320–322. - PubMed
-
- Danpure C, Jennings P. Peroxisosomal alanine glyoxylate aminotransferase deficiency in primary hyperoxaluria type I. FEBS. 1986;201:20–24. - PubMed
-
- Kamoun A, Lakhoua R. Endstage renal disease of the Tunisian child: epidemiology, etiologies, and outcome. Pediatr Nephrol. 1996;10:479–482. - PubMed
-
- Al-Eisa AA, Samham M, Naseef M. End-stage renal disease in Kuwaiti children: an 8 year experience. Transplant Proc. 2004;36:1788–1791. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- RC2 HL102926/HL/NHLBI NIH HHS/United States
- HL-102924/HL/NHLBI NIH HHS/United States
- RC2 HL102924/HL/NHLBI NIH HHS/United States
- HL-102926/HL/NHLBI NIH HHS/United States
- HL-102925/HL/NHLBI NIH HHS/United States
- RC2 HL103010/HL/NHLBI NIH HHS/United States
- HL-102923/HL/NHLBI NIH HHS/United States
- RC2 HL102923/HL/NHLBI NIH HHS/United States
- UC2 HL102926/HL/NHLBI NIH HHS/United States
- UC2 HL103010/HL/NHLBI NIH HHS/United States
- HL-103010/HL/NHLBI NIH HHS/United States
- UC2 HL102923/HL/NHLBI NIH HHS/United States
- UC2 HL102924/HL/NHLBI NIH HHS/United States
- RC2 HL102925/HL/NHLBI NIH HHS/United States
- UC2 HL102925/HL/NHLBI NIH HHS/United States
- K08 DK095994/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
