

The mechanosensory structure of the hair cell requires clarin-1, a protein encoded by Usher syndrome III causative gene
- PMID: 22787034
- PMCID: PMC3422646
- DOI: 10.1523/JNEUROSCI.0311-12.2012
The mechanosensory structure of the hair cell requires clarin-1, a protein encoded by Usher syndrome III causative gene
Abstract
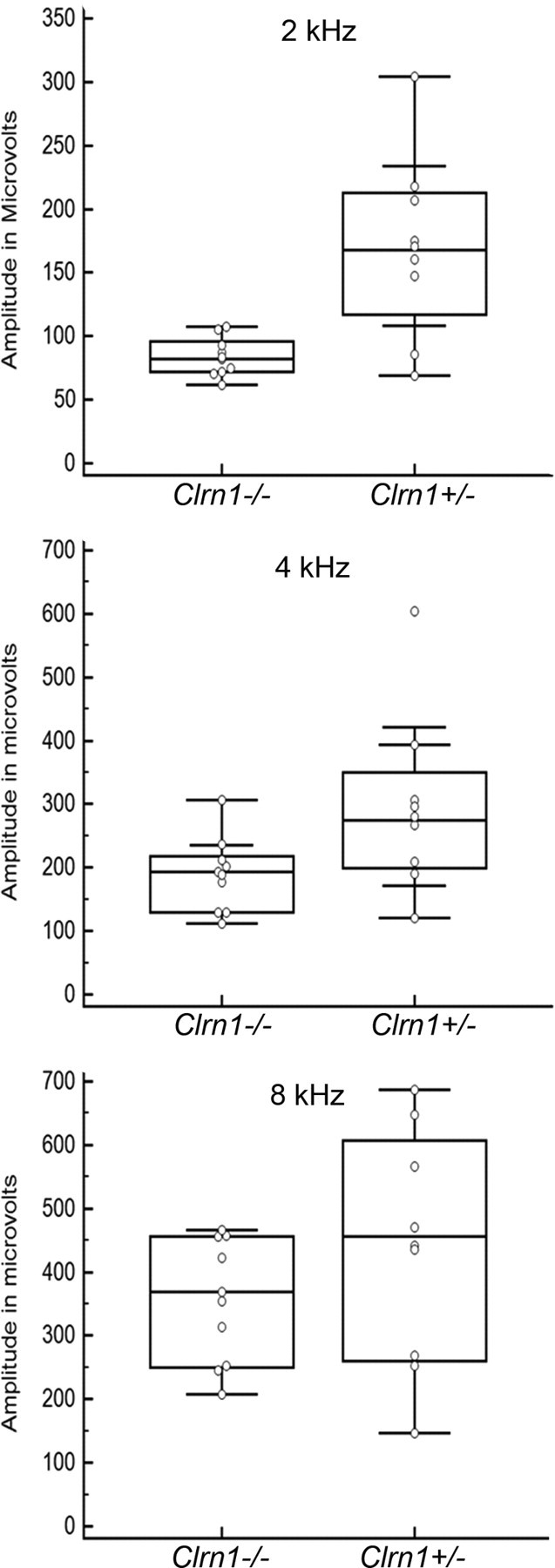
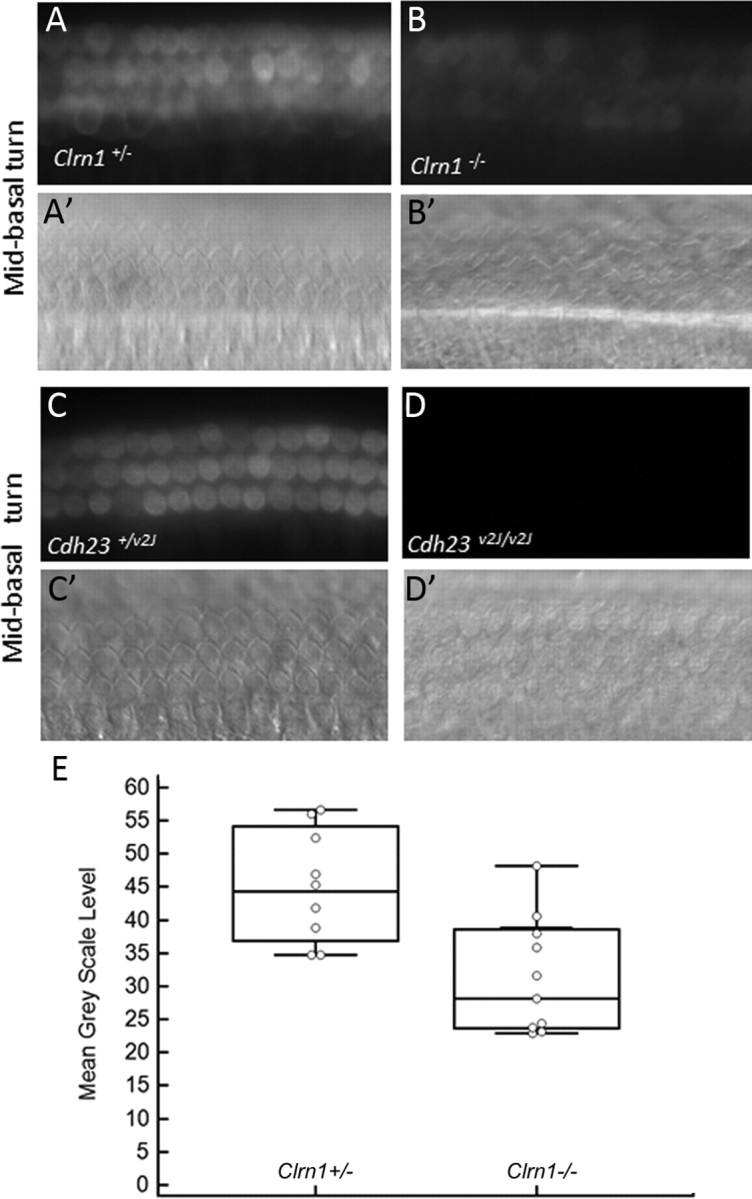
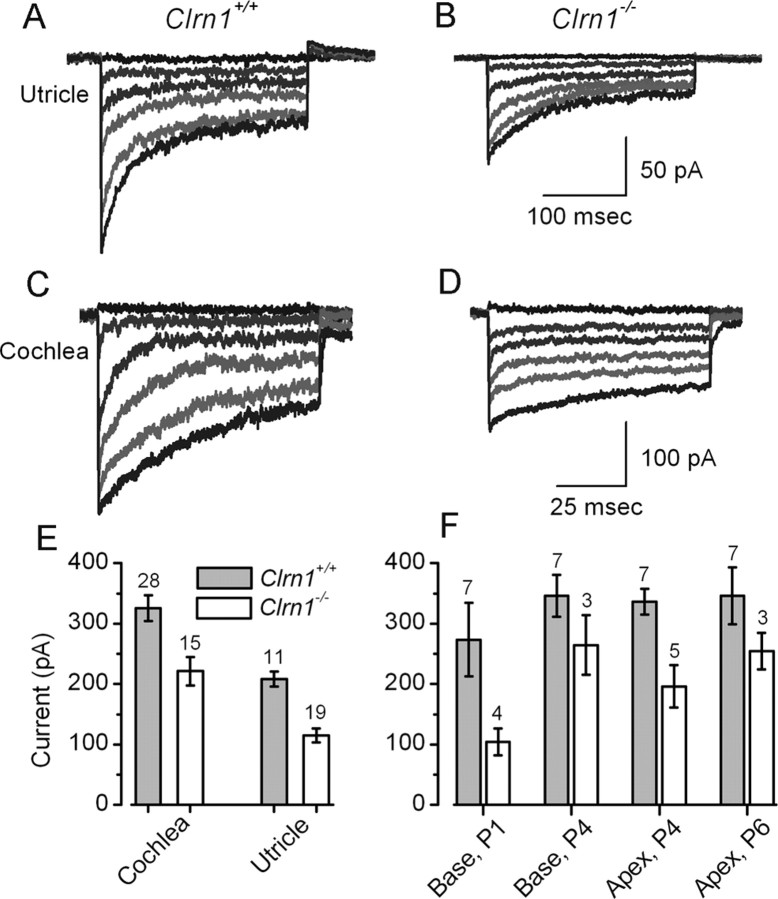
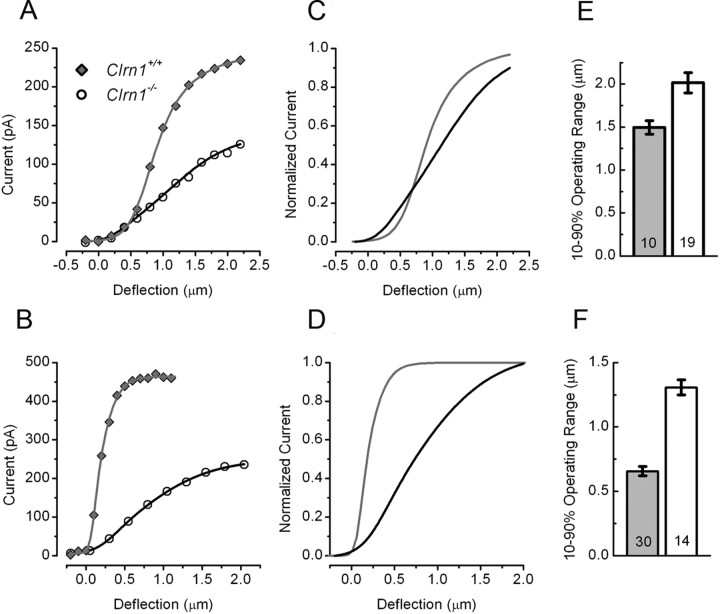
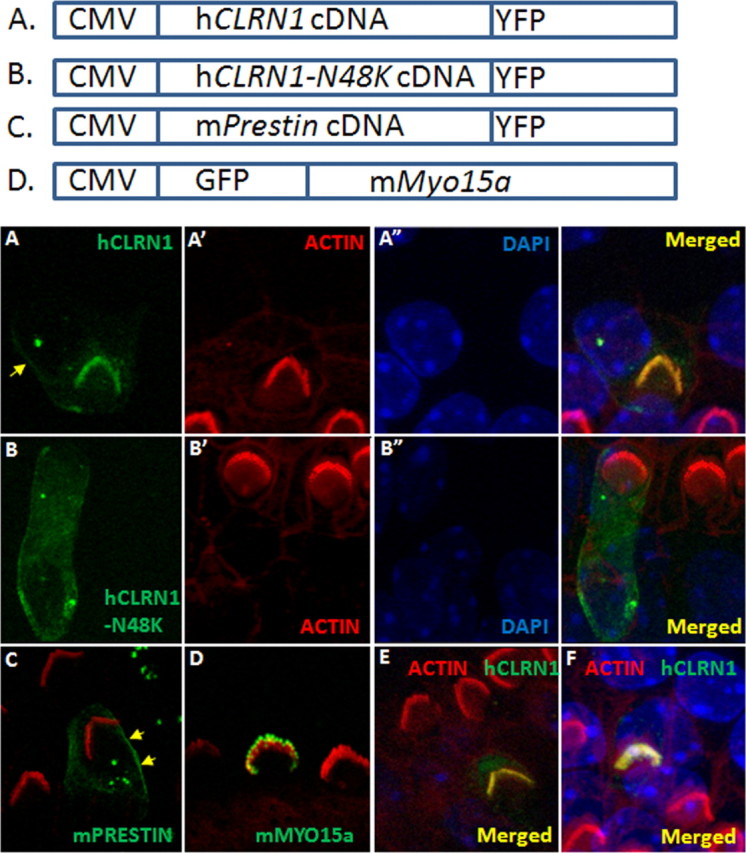
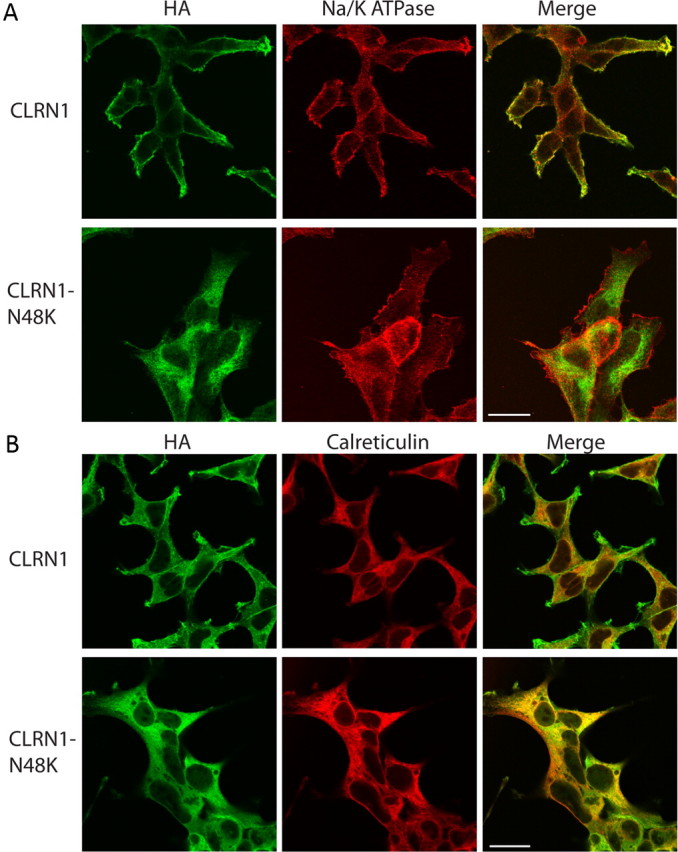
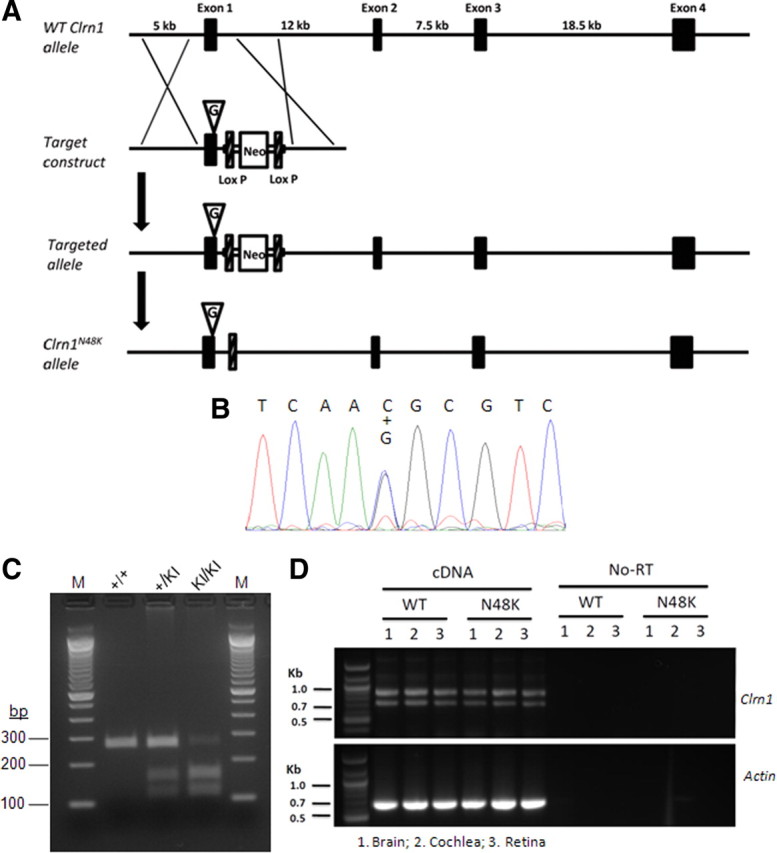
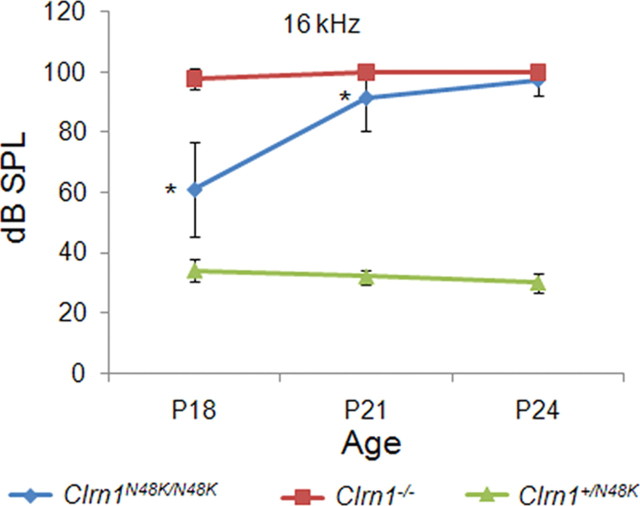
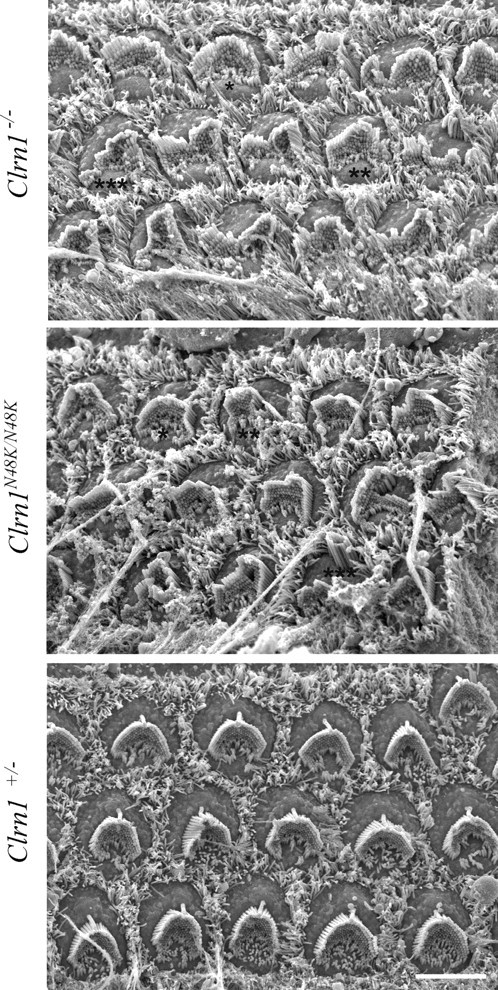
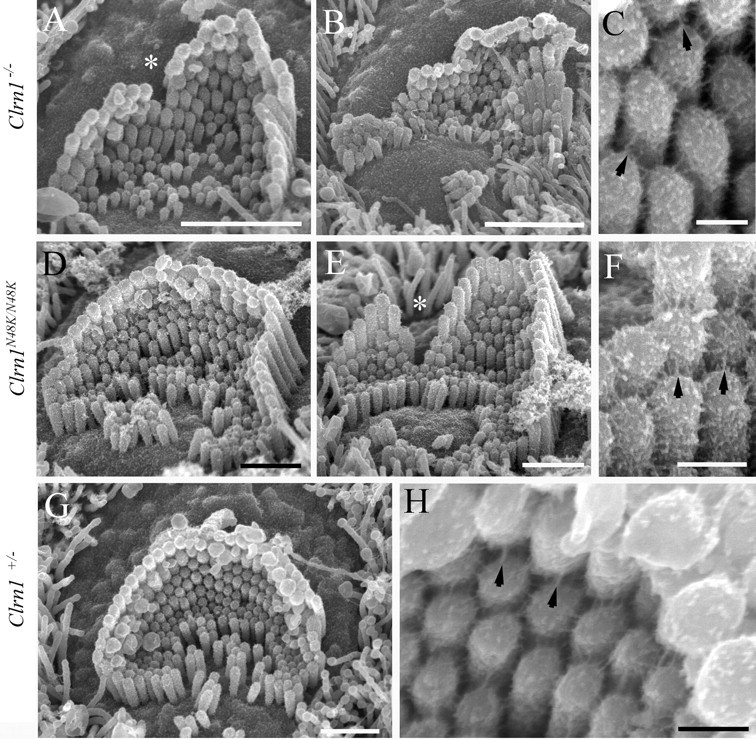
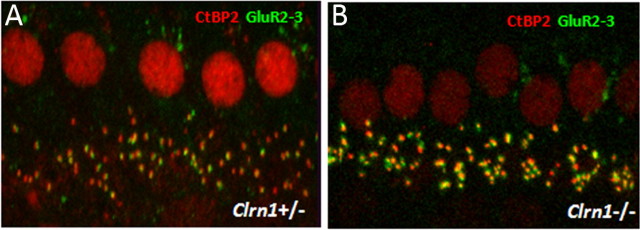
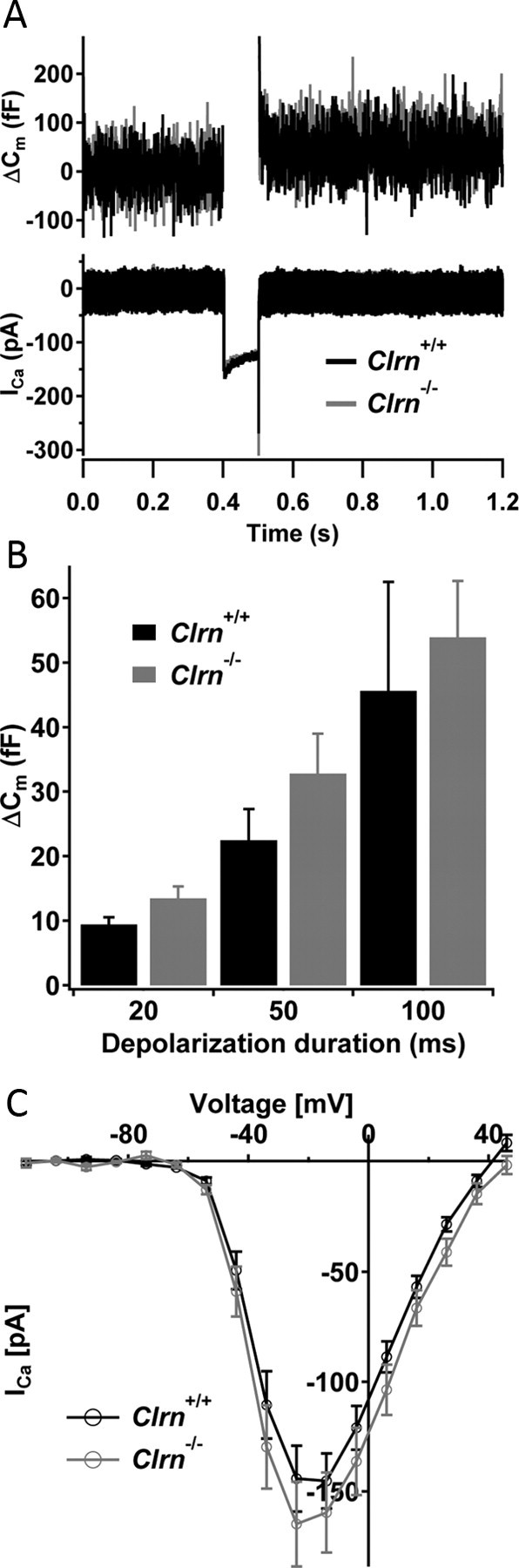
Mutation in the clarin-1 gene (Clrn1) results in loss of hearing and vision in humans (Usher syndrome III), but the role of clarin-1 in the sensory hair cells is unknown. Clarin-1 is predicted to be a four transmembrane domain protein similar to members of the tetraspanin family. Mice carrying null mutation in the clarin-1 gene (Clrn1(-/-)) show loss of hair cell function and a possible defect in ribbon synapse. We investigated the role of clarin-1 using various in vitro and in vivo approaches. We show by immunohistochemistry and patch-clamp recordings of Ca(2+) currents and membrane capacitance from inner hair cells that clarin-1 is not essential for formation or function of ribbon synapse. However, reduced cochlear microphonic potentials, FM1-43 [N-(3-triethylammoniumpropyl)-4-(4-(dibutylamino)styryl) pyridinium dibromide] loading, and transduction currents pointed to diminished cochlear hair bundle function in Clrn1(-/-) mice. Electron microscopy of cochlear hair cells revealed loss of some tall stereocilia and gaps in the v-shaped bundle, although tip links and staircase arrangement of stereocilia were not primarily affected by Clrn1(-/-) mutation. Human clarin-1 protein expressed in transfected mouse cochlear hair cells localized to the bundle; however, the pathogenic variant p.N48K failed to localize to the bundle. The mouse model generated to study the in vivo consequence of p.N48K in clarin-1 (Clrn1(N48K)) supports our in vitro and Clrn1(-/-) mouse data and the conclusion that CLRN1 is an essential hair bundle protein. Furthermore, the ear phenotype in the Clrn1(N48K) mouse suggests that it is a valuable model for ear disease in CLRN1(N48K), the most prevalent Usher syndrome III mutation in North America.
Figures
References
-
- Adato A, Vreugde S, Joensuu T, Avidan N, Hamalainen R, Belenkiy O, Olender T, Bonne-Tamir B, Ben-Asher E, Espinos C, Millán JM, Lehesjoki AE, Flannery JG, Avraham KB, Pietrokovski S, Sankila EM, Beckmann JS, Lancet D. USH3A transcripts encode clarin-1, a four-transmembranedomain protein with a possible role in sensory synapses. Eur J Hum Genet. 2002;10:339–350. - PubMed
-
- Ahmed ZM, Goodyear R, Riazuddin S, Lagziel A, Legan PK, Behra M, Burgess SM, Lilley KS, Wilcox ER, Riazuddin S, Griffith AJ, Frolenkov GI, Belyantseva IA, Richardson GP, Friedman TB. The tip-link antigen, a protein associated with the transduction complex of sensory hair cells, is protocadherin-15. J Neurosci. 2006;26:7022–7034. - PMC - PubMed
-
- Alagramam KN, Murcia CL, Kwon HY, Pawlowski KS, Wright CG, Woychik RP. The mouse Ames waltzer hearing-loss mutant is caused by mutation of Pcdh15, a novel protocadherin gene. Nat Genet. 2001;27:99–102. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous