

Aldosterone inactivates the endothelin-B receptor via a cysteinyl thiol redox switch to decrease pulmonary endothelial nitric oxide levels and modulate pulmonary arterial hypertension
- PMID: 22787113
- PMCID: PMC3534848
- DOI: 10.1161/CIRCULATIONAHA.112.094722
Aldosterone inactivates the endothelin-B receptor via a cysteinyl thiol redox switch to decrease pulmonary endothelial nitric oxide levels and modulate pulmonary arterial hypertension
Abstract
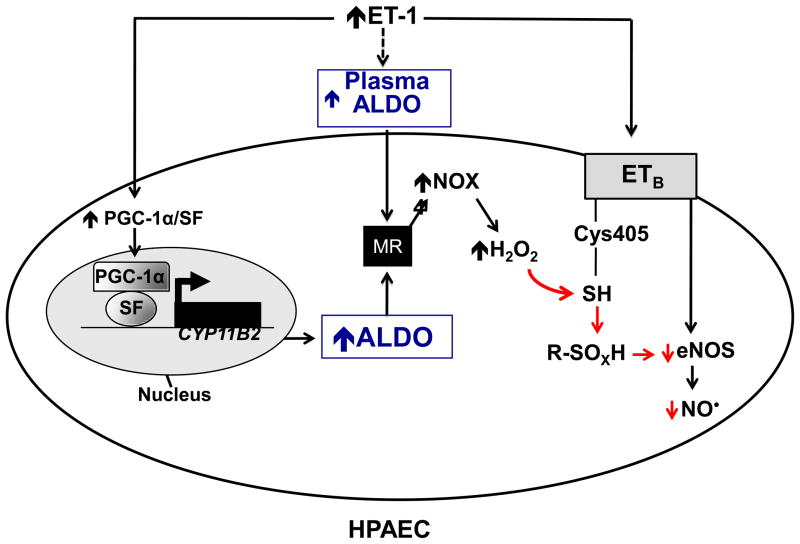
Background: Pulmonary arterial hypertension (PAH) is characterized, in part, by decreased endothelial nitric oxide (NO(·)) production and elevated levels of endothelin-1. Endothelin-1 is known to stimulate endothelial nitric oxide synthase (eNOS) via the endothelin-B receptor (ET(B)), suggesting that this signaling pathway is perturbed in PAH. Endothelin-1 also stimulates adrenal aldosterone synthesis; in systemic blood vessels, hyperaldosteronism induces vascular dysfunction by increasing endothelial reactive oxygen species generation and decreasing NO(·) levels. We hypothesized that aldosterone modulates PAH by disrupting ET(B)-eNOS signaling through a mechanism involving increased pulmonary endothelial oxidant stress.
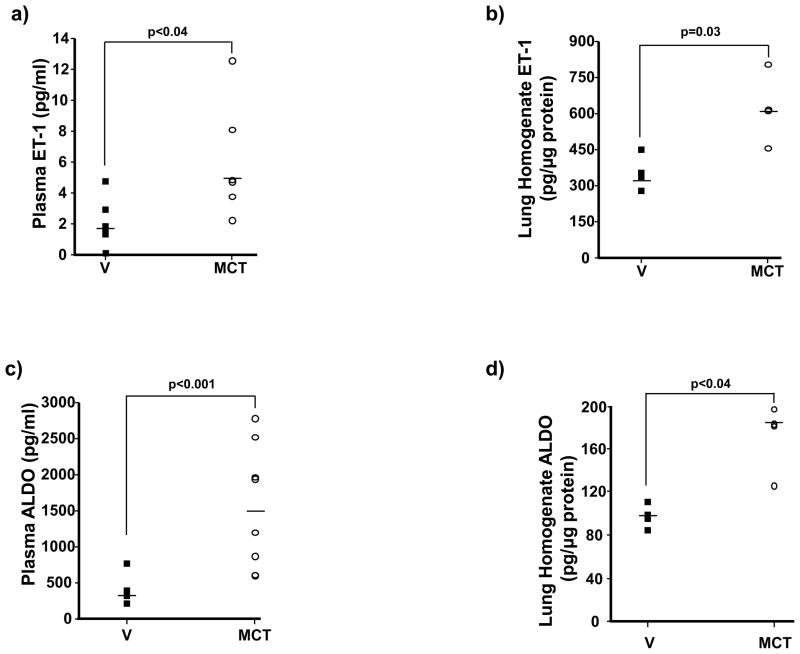
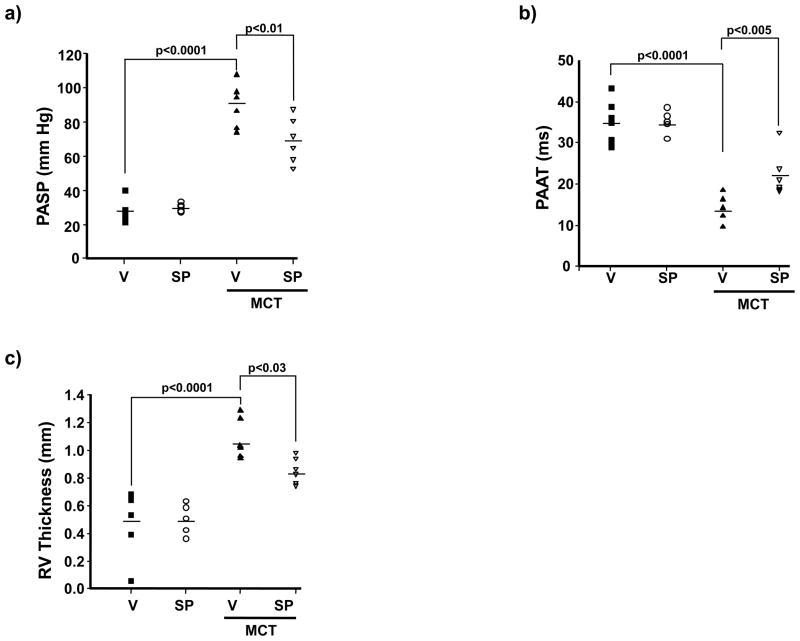
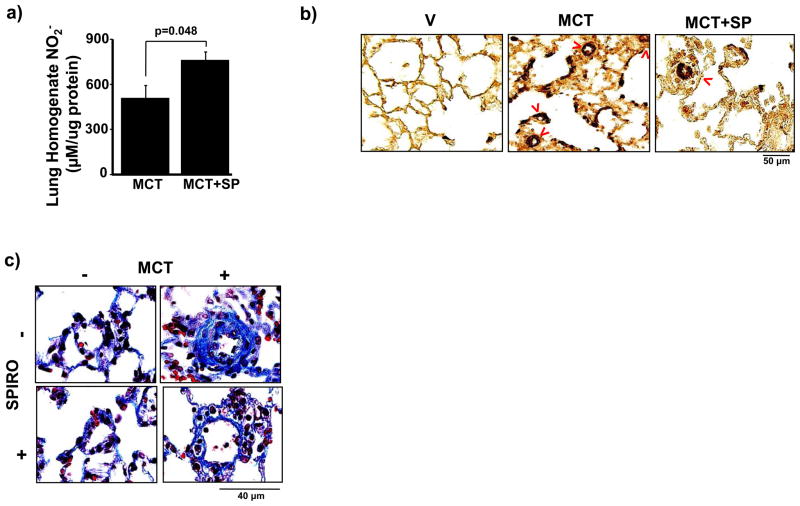
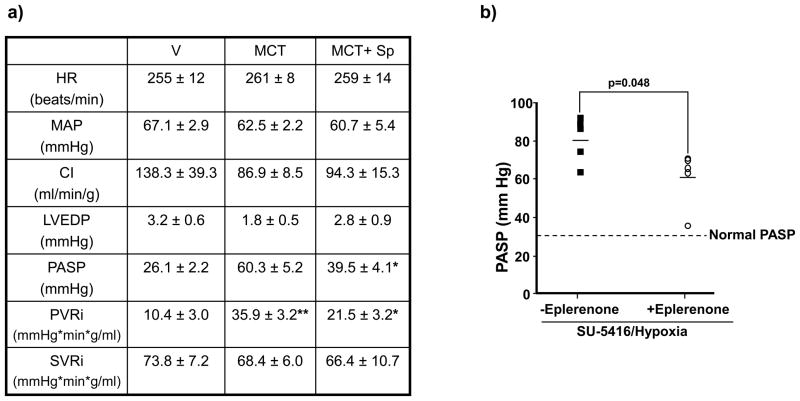
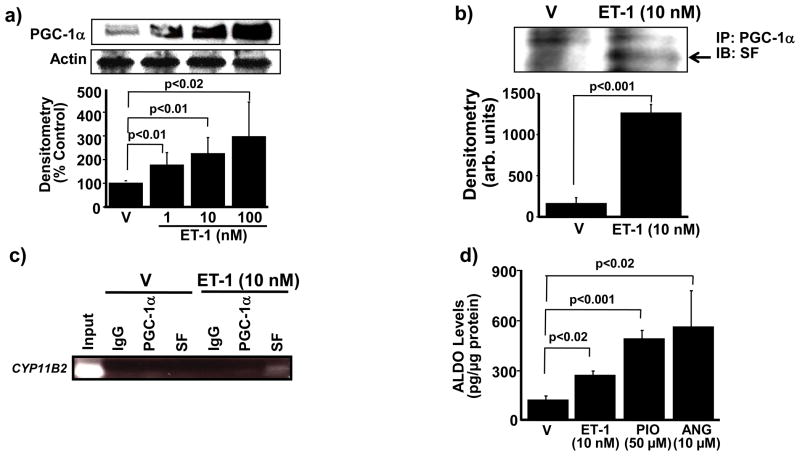
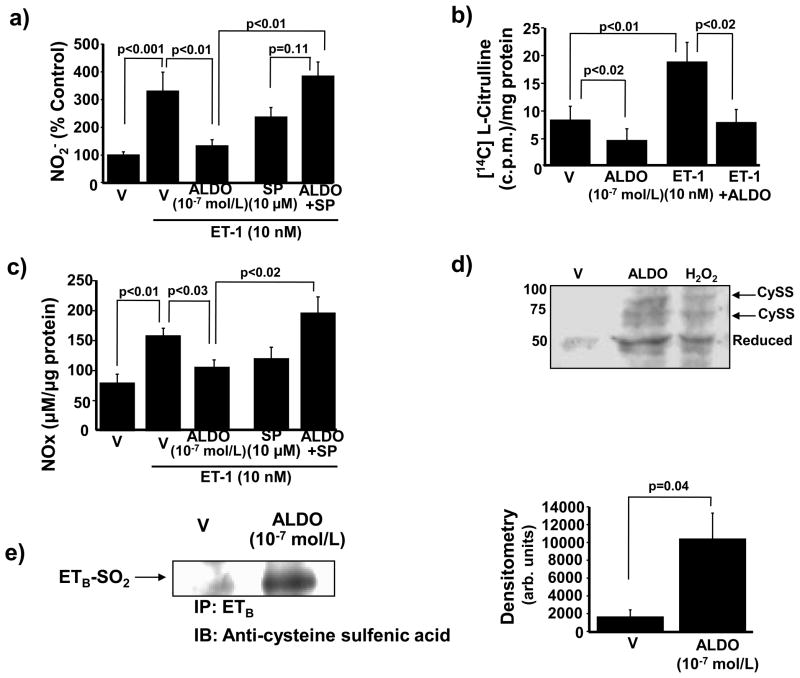
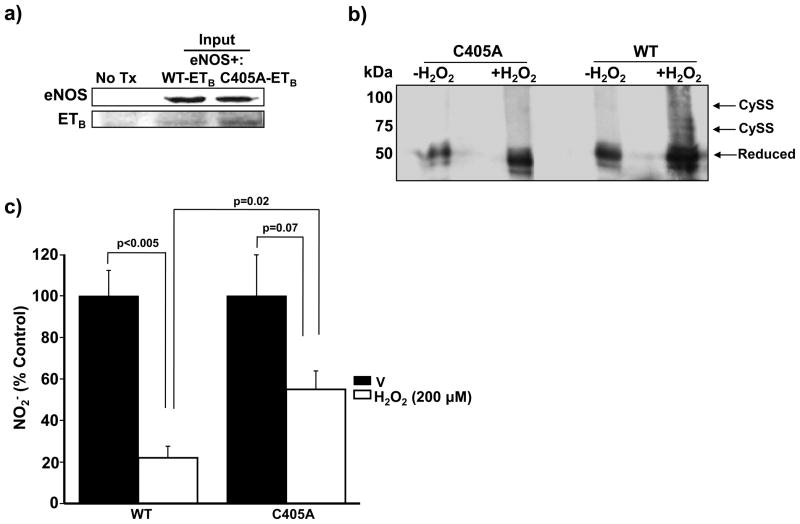
Methods and results: In rats with PAH, elevated endothelin-1 levels were associated with elevated aldosterone levels in plasma and lung tissue and decreased lung NO(·) metabolites in the absence of left-sided heart failure. In human pulmonary artery endothelial cells, endothelin-1 increased aldosterone levels via peroxisome proliferator-activated receptor gamma coactivator-1α/steroidogenesis factor-1-dependent upregulation of aldosterone synthase. Aldosterone also increased reactive oxygen species production, which oxidatively modified cysteinyl thiols in the eNOS-activating region of ET(B) to decrease endothelin-1-stimulated eNOS activity. Substitution of ET(B)-Cys405 with alanine improved ET(B)-dependent NO(·) synthesis under conditions of oxidant stress, confirming that Cys405 is a redox-sensitive thiol that is necessary for ET(B)-eNOS signaling. In human pulmonary artery endothelial cells, mineralocorticoid receptor antagonism with spironolactone decreased aldosterone-mediated reactive oxygen species generation and restored ET(B)-dependent NO(·) production. Spironolactone or eplerenone prevented or reversed pulmonary vascular remodeling and improved cardiopulmonary hemodynamics in 2 animal models of PAH in vivo.
Conclusions: Our findings demonstrate that aldosterone modulates an ET(B) cysteinyl thiol redox switch to decrease pulmonary endothelium-derived NO(·) and promote PAH.
Conflict of interest statement
None.
Figures
References
-
- Farber HW, Loscalzo J. Pulmonary arterial hypertension. N Engl J Med. 2004;351:1655–1665. - PubMed
-
- Michelakis ED. The role of the NO axis and its therapeutic implications in pulmonary arterial hypertension. Heart Fail Rev. 2003;8:5–21. - PubMed
-
- Weber KT. Aldosterone in congestive heart failure. N Engl J Med. 2001;345:1689–1697. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- U01 HL108630/HL/NHLBI NIH HHS/United States
- U54 HL070819/HL/NHLBI NIH HHS/United States
- P01 HL048743/HL/NHLBI NIH HHS/United States
- R01 HL061795/HL/NHLBI NIH HHS/United States
- K08 HL096834/HL/NHLBI NIH HHS/United States
- HL108630/HL/NHLBI NIH HHS/United States
- K08 HL111207/HL/NHLBI NIH HHS/United States
- HL48743/HL/NHLBI NIH HHS/United States
- R37 HL061795/HL/NHLBI NIH HHS/United States
- HL107192/HL/NHLBI NIH HHS/United States
- HL105301/HL/NHLBI NIH HHS/United States
- P50 HL107192/HL/NHLBI NIH HHS/United States
- T32 HL007604/HL/NHLBI NIH HHS/United States
- HL61795/HL/NHLBI NIH HHS/United States
- HL070819/HL/NHLBI NIH HHS/United States
- R01 HL105301/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
