

Biomolecular dynamics: order-disorder transitions and energy landscapes
- PMID: 22790780
- PMCID: PMC3695400
- DOI: 10.1088/0034-4885/75/7/076601
Biomolecular dynamics: order-disorder transitions and energy landscapes
Abstract
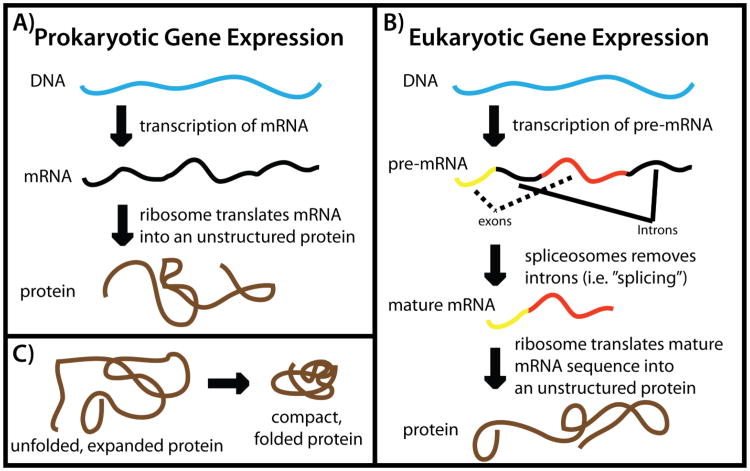
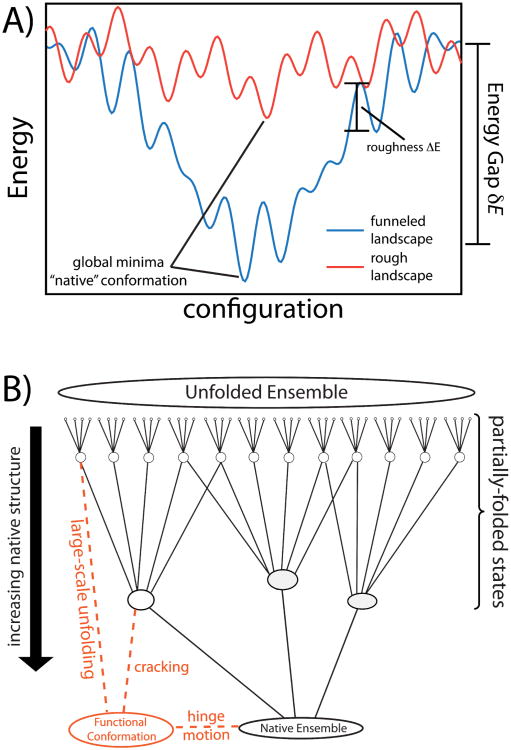
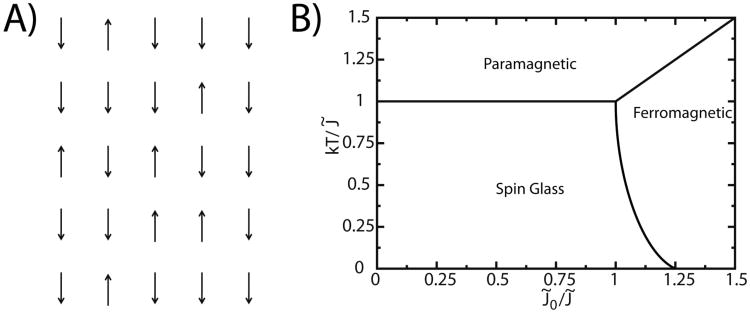
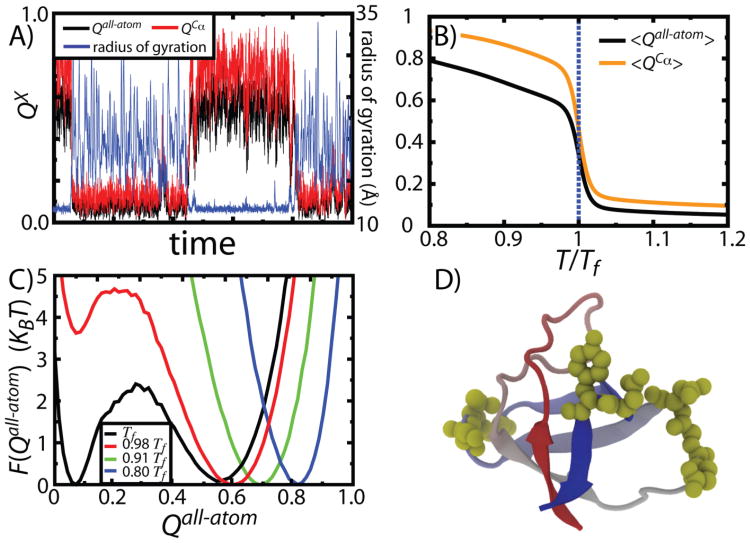
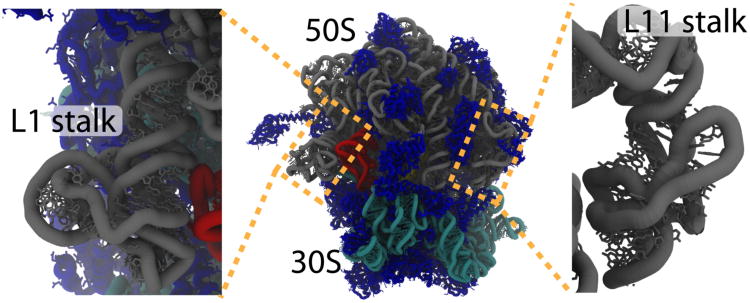
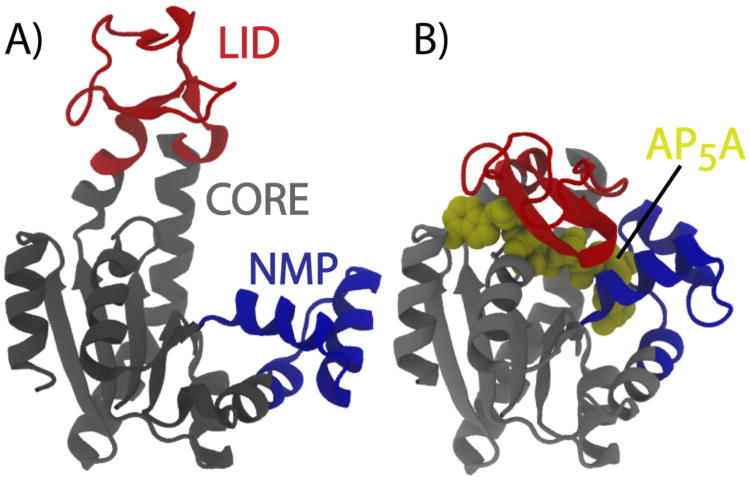
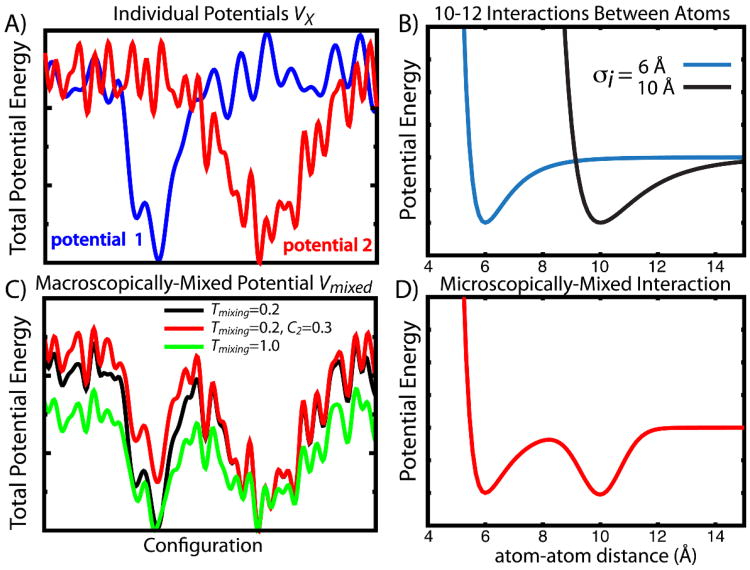
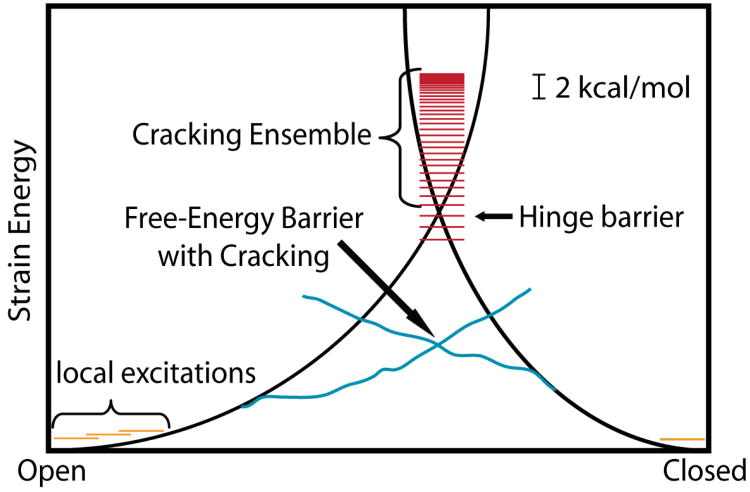
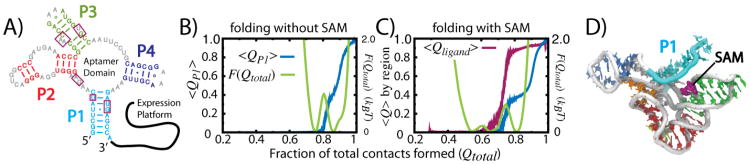
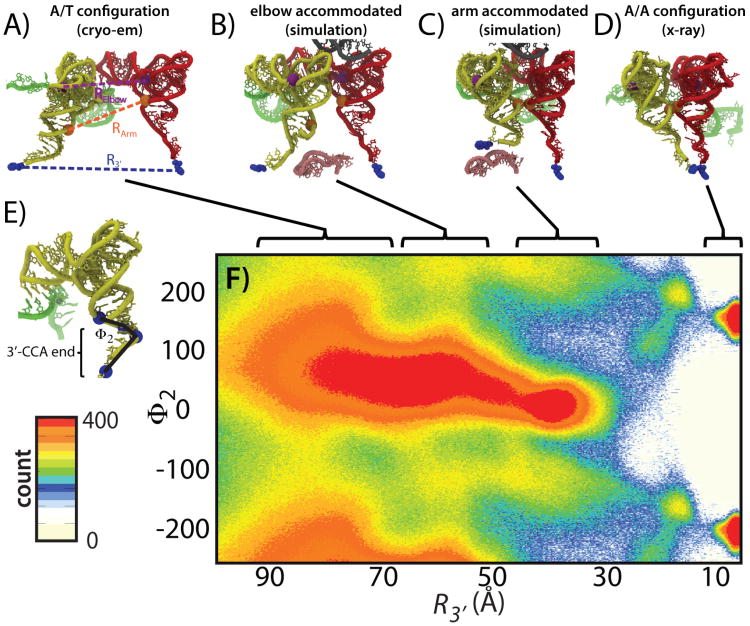
While the energy landscape theory of protein folding is now a widely accepted view for understanding how relatively weak molecular interactions lead to rapid and cooperative protein folding, such a framework must be extended to describe the large-scale functional motions observed in molecular machines. In this review, we discuss (1) the development of the energy landscape theory of biomolecular folding, (2) recent advances toward establishing a consistent understanding of folding and function and (3) emerging themes in the functional motions of enzymes, biomolecular motors and other biomolecular machines. Recent theoretical, computational and experimental lines of investigation have provided a very dynamic picture of biomolecular motion. In contrast to earlier ideas, where molecular machines were thought to function similarly to macroscopic machines, with rigid components that move along a few degrees of freedom in a deterministic fashion, biomolecular complexes are only marginally stable. Since the stabilizing contribution of each atomic interaction is on the order of the thermal fluctuations in solution, the rigid body description of molecular function must be revisited. An emerging theme is that functional motions encompass order-disorder transitions and structural flexibility provides significant contributions to the free energy. In this review, we describe the biological importance of order-disorder transitions and discuss the statistical-mechanical foundation of theoretical approaches that can characterize such transitions.
Figures
Similar articles
-
Analysis of functional motions in Brownian molecular machines with an efficient block normal mode approach: myosin-II and Ca2+ -ATPase.Biophys J. 2004 Feb;86(2):743-63. doi: 10.1016/S0006-3495(04)74152-1. Biophys J. 2004. PMID: 14747312 Free PMC article.
-
Entropic control of the free-energy landscape of an archetypal biomolecular machine.Proc Natl Acad Sci U S A. 2023 May 23;120(21):e2220591120. doi: 10.1073/pnas.2220591120. Epub 2023 May 15. Proc Natl Acad Sci U S A. 2023. PMID: 37186858 Free PMC article.
-
Brownian ratchet models of molecular motors.Cell Biochem Biophys. 2003;38(2):191-214. doi: 10.1385/CBB:38:2:191. Cell Biochem Biophys. 2003. PMID: 12777714 Review.
-
Mesoscale modeling of molecular machines: cyclic dynamics and hydrodynamical fluctuations.Phys Rev E Stat Nonlin Soft Matter Phys. 2008 May;77(5 Pt 1):050901. doi: 10.1103/PhysRevE.77.050901. Epub 2008 May 7. Phys Rev E Stat Nonlin Soft Matter Phys. 2008. PMID: 18643015
-
Protein structural transitions and their functional role.Philos Trans A Math Phys Eng Sci. 2005 Feb 15;363(1827):331-55; discussion 355-6. doi: 10.1098/rsta.2004.1496. Philos Trans A Math Phys Eng Sci. 2005. PMID: 15664887 Review.
Cited by
-
Critical phenomena in the temperature-pressure-crowding phase diagram of a protein.Phys Rev X. 2019 Oct-Dec;9(4):041035. doi: 10.1103/physrevx.9.041035. Epub 2019 Nov 18. Phys Rev X. 2019. PMID: 32642303 Free PMC article.
-
The Role of Electrostatics and Folding Kinetics on the Thermostability of Homologous Cold Shock Proteins.J Chem Inf Model. 2020 Feb 24;60(2):546-561. doi: 10.1021/acs.jcim.9b00797. Epub 2020 Jan 17. J Chem Inf Model. 2020. PMID: 31910002 Free PMC article.
-
Dramatic Shape Changes Occur as Cytochrome c Folds.J Phys Chem B. 2020 Sep 24;124(38):8240-8248. doi: 10.1021/acs.jpcb.0c05802. Epub 2020 Sep 9. J Phys Chem B. 2020. PMID: 32840372 Free PMC article.
-
Intrinsic Dynamics of the ClpXP Proteolytic Machine Using Elastic Network Models.ACS Omega. 2023 Feb 14;8(8):7302-7318. doi: 10.1021/acsomega.2c04347. eCollection 2023 Feb 28. ACS Omega. 2023. PMID: 36873006 Free PMC article.
-
Binding of interferon reduces the force of unfolding for interferon receptor 1.PLoS One. 2017 Apr 12;12(4):e0175413. doi: 10.1371/journal.pone.0175413. eCollection 2017. PLoS One. 2017. PMID: 28403186 Free PMC article.
References
-
- Cech TR. Self-splicing of group-I introns. Annu Rev Biochem. 1990 Jan;59:543–568. - PubMed
-
- Mcclain WH, Guerriertakada C, Altman S. Model substrates for an RNA enzyme. Science. 1987 Jan;238(4826):527–530. - PubMed
-
- Yusupov MM, Yusupova GZ, Baucom A, Lieberman K, Earnest TN, Cate JH, Noller HF. Crystal structure of the ribosome at 5.5 a resolution. Science. 2001 May;92(5518):883–96. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources