

Predicting signatures of "synthetic associations" and "natural associations" from empirical patterns of human genetic variation
- PMID: 22792059
- PMCID: PMC3390358
- DOI: 10.1371/journal.pcbi.1002600
Predicting signatures of "synthetic associations" and "natural associations" from empirical patterns of human genetic variation
Abstract
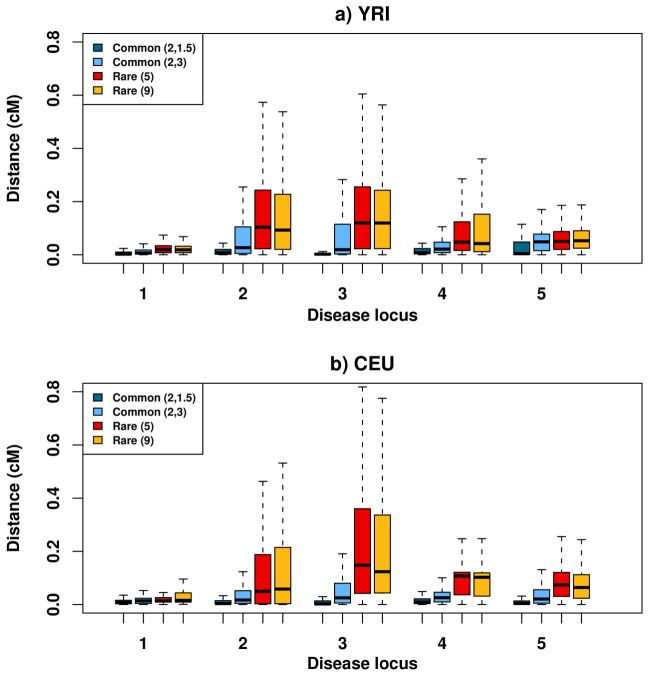
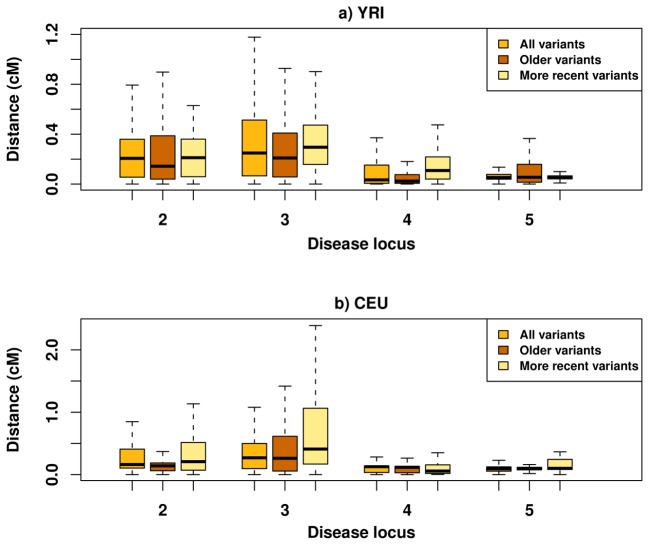
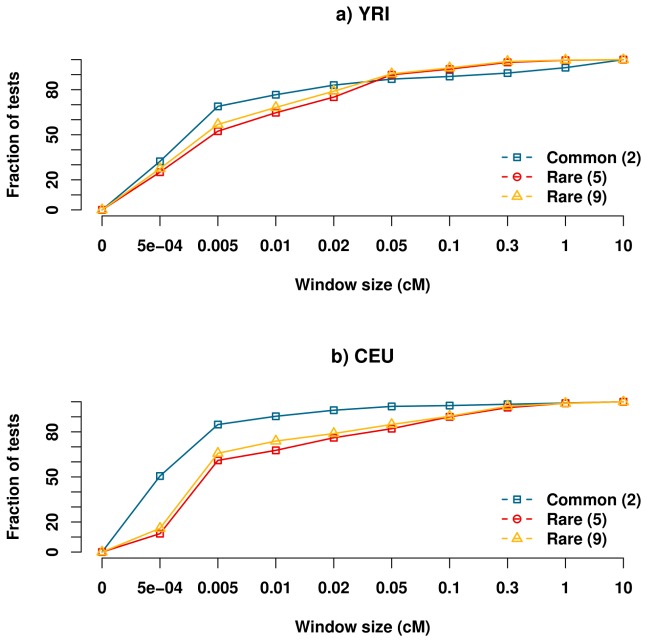
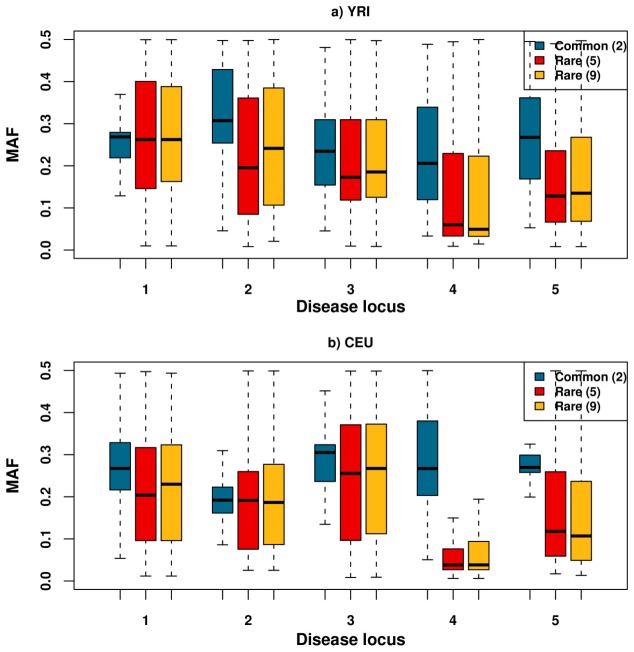
Genome-wide association studies (GWAS) have in recent years discovered thousands of associated markers for hundreds of phenotypes. However, associated loci often only explain a relatively small fraction of heritability and the link between association and causality has yet to be uncovered for most loci. Rare causal variants have been suggested as one scenario that may partially explain these shortcomings. Specifically, Dickson et al. recently reported simulations of rare causal variants that lead to association signals of common, tag single nucleotide polymorphisms, dubbed "synthetic associations". However, an open question is what practical implications synthetic associations have for GWAS. Here, we explore the signatures exhibited by such "synthetic associations" and their implications based on patterns of genetic variation observed in human populations, thus accounting for human evolutionary history -a force disregarded in previous simulation studies. This is made possible by human population genetic data from HapMap 3 consisting of both resequencing and array-based genotyping data for the same set of individuals from multiple populations. We report that synthetic associations tend to be further away from the underlying risk alleles compared to "natural associations" (i.e. associations due to underlying common causal variants), but to a much lesser extent than previously predicted, with both the age and the effect size of the risk allele playing a part in this phenomenon. We find that while a synthetic association has a lower probability of capturing causal variants within its linkage disequilibrium block, sequencing around the associated variant need not extend substantially to have a high probability of capturing at least one causal variant. We also show that the minor allele frequency of synthetic associations is lower than of natural associations for most, but not all, loci that we explored. Finally, we find the variance in associated allele frequency to be a potential indicator of synthetic associations.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Pritchard JK, Cox NJ. The allelic architecture of human disease genes: common disease-common variant…or not? Hum Mol Genet. 2002;11:2417–2423. - PubMed
-
- Reich DE, Lander ES. On the allelic spectrum of human disease. Trends Genet. 2001;17:502–510. - PubMed
-
- Frazer KA, Murray SS, Schork NJ, Topol EJ. Human genetic variation and its contribution to complex traits. Nat Rev Genet. 2009;10:241–251. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
