

Mitochondrial energy metabolism and redox signaling in brain aging and neurodegeneration
- PMID: 22793257
- PMCID: PMC3887431
- DOI: 10.1089/ars.2012.4774
Mitochondrial energy metabolism and redox signaling in brain aging and neurodegeneration
Abstract
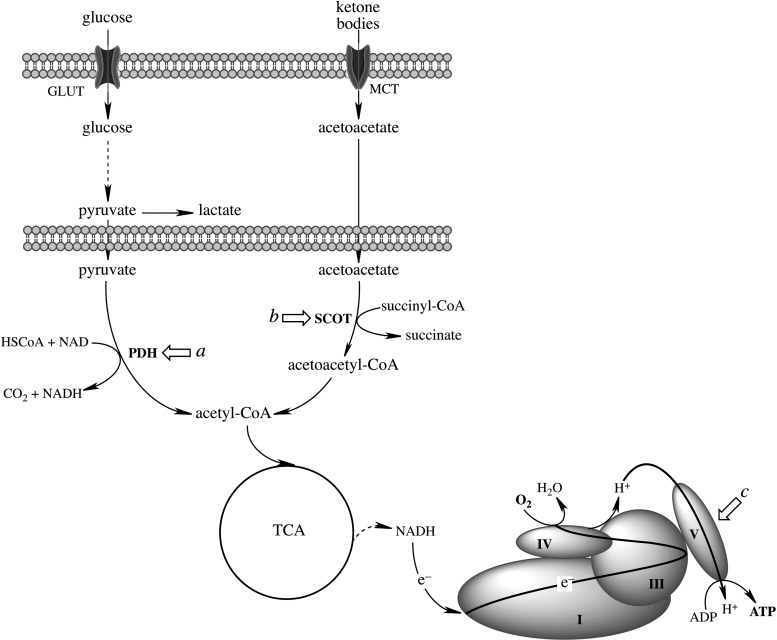
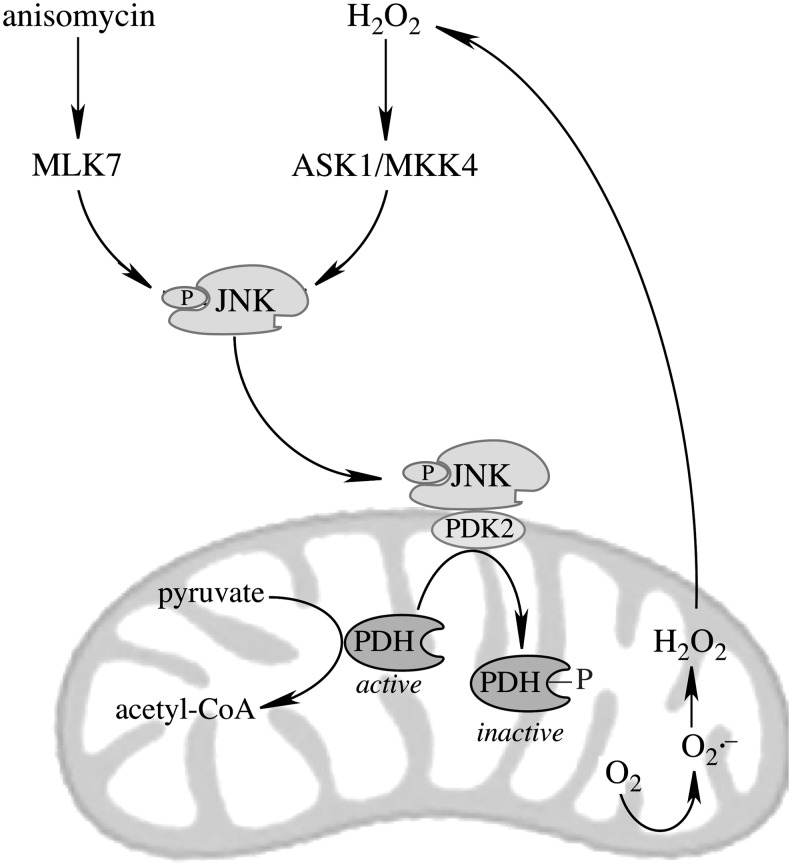
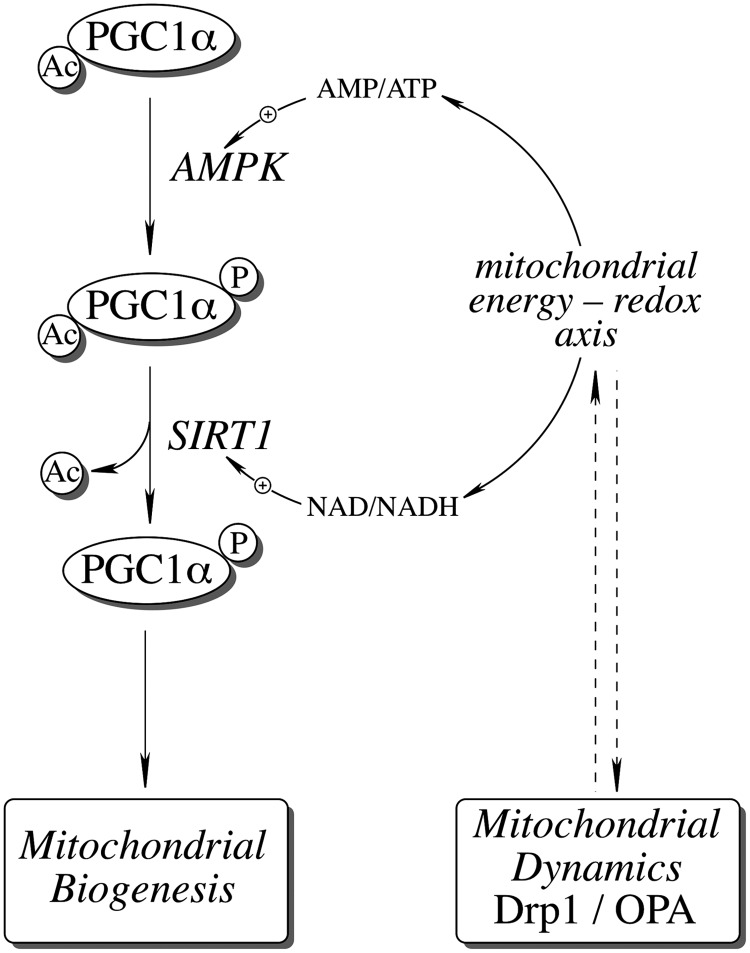
Significance: The mitochondrial energy-transducing capacity is essential for the maintenance of neuronal function, and the impairment of energy metabolism and redox homeostasis is a hallmark of brain aging, which is particularly accentuated in the early stages of neurodegenerative diseases.
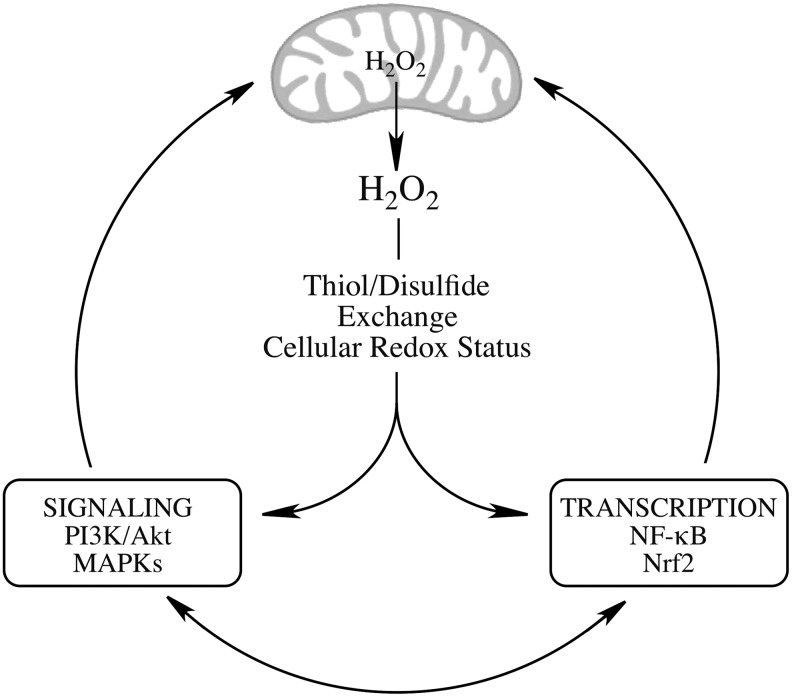
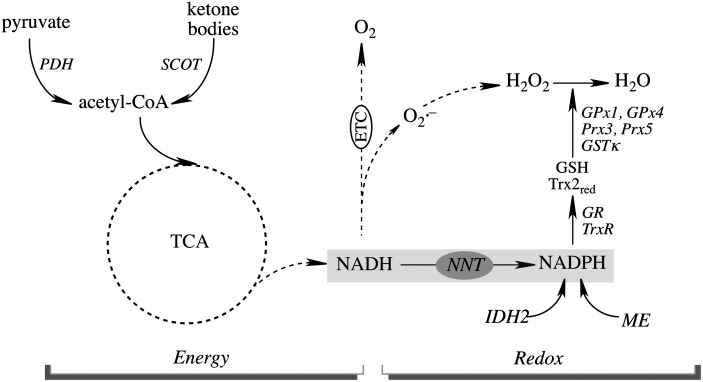
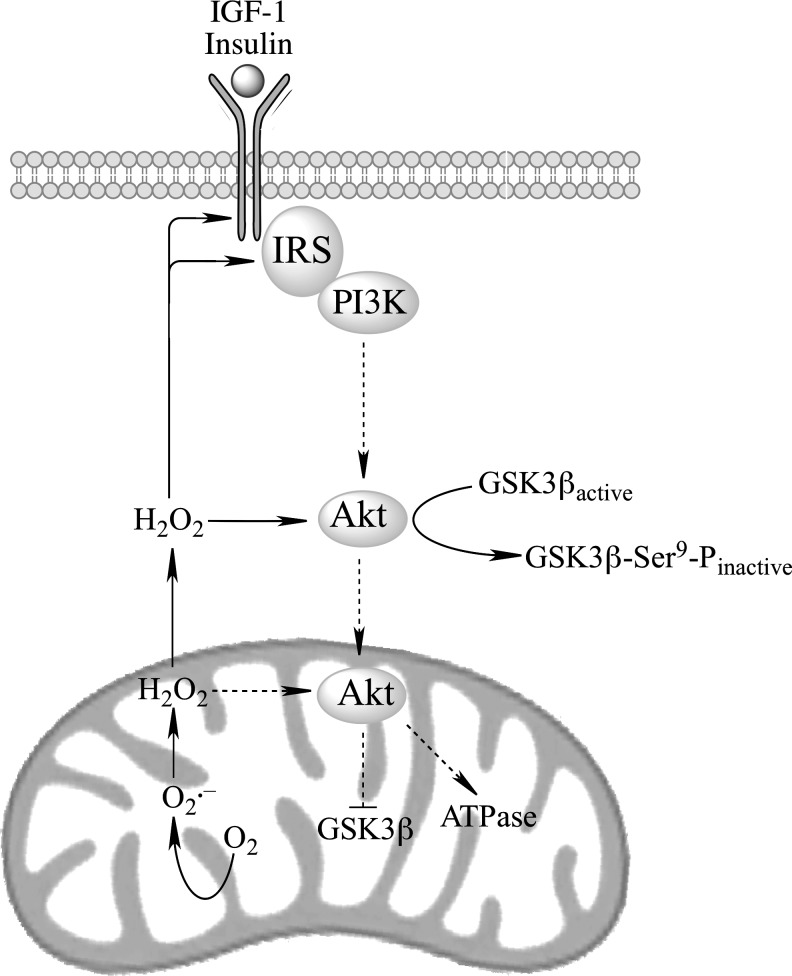
Recent advances: The communications between mitochondria and the rest of the cell by energy- and redox-sensitive signaling establish a master regulatory device that controls cellular energy levels and the redox environment. Impairment of this regulatory devise is critical for aging and the early stages of neurodegenerative diseases.
Critical issues: This review focuses on a coordinated metabolic network-cytosolic signaling, transcriptional regulation, and mitochondrial function-that controls the cellular energy levels and redox status as well as factors which impair this metabolic network during brain aging and neurodegeneration.
Future directions: Characterization of mitochondrial function and mitochondria-cytosol communications will provide pivotal opportunities for identifying targets and developing new strategies aimed at restoring the mitochondrial energy-redox axis that is compromised in brain aging and neurodegeneration.
Figures
Similar articles
-
The energy-redox axis in aging and age-related neurodegeneration.Adv Drug Deliv Rev. 2009 Nov 30;61(14):1283-98. doi: 10.1016/j.addr.2009.07.015. Epub 2009 Aug 27. Adv Drug Deliv Rev. 2009. PMID: 19716388 Free PMC article. Review.
-
Sirtuins and Their Roles in Brain Aging and Neurodegenerative Disorders.Neurochem Res. 2017 Mar;42(3):876-890. doi: 10.1007/s11064-016-2110-y. Epub 2016 Nov 24. Neurochem Res. 2017. PMID: 27882448 Free PMC article. Review.
-
Proteotoxicity and mitochondrial dynamics in aging diabetic brain.Pharmacol Res. 2020 Sep;159:104948. doi: 10.1016/j.phrs.2020.104948. Epub 2020 May 22. Pharmacol Res. 2020. PMID: 32450345 Review.
-
Mitochondrial turnover and homeostasis in ageing and neurodegeneration.FEBS Lett. 2020 Aug;594(15):2370-2379. doi: 10.1002/1873-3468.13802. Epub 2020 Jun 1. FEBS Lett. 2020. PMID: 32350855 Review.
-
Ground control to major TOM: mitochondria-nucleus communication.FEBS J. 2017 Jan;284(2):196-210. doi: 10.1111/febs.13778. Epub 2016 Jul 4. FEBS J. 2017. PMID: 27283924 Review.
Cited by
-
Potential role of cannabidiol in Parkinson's disease by targeting the WNT/β-catenin pathway, oxidative stress and inflammation.Aging (Albany NY). 2021 Apr 13;13(7):10796-10813. doi: 10.18632/aging.202951. Epub 2021 Apr 13. Aging (Albany NY). 2021. PMID: 33848261 Free PMC article. Review.
-
New insights into the interplay between autophagy and oxidative and endoplasmic reticulum stress in neuronal cell death and survival.Front Cell Dev Biol. 2022 Sep 16;10:994037. doi: 10.3389/fcell.2022.994037. eCollection 2022. Front Cell Dev Biol. 2022. PMID: 36187470 Free PMC article. Review.
-
Cellular Specificity and Inter-cellular Coordination in the Brain Bioenergetic System: Implications for Aging and Neurodegeneration.Front Physiol. 2020 Jan 8;10:1531. doi: 10.3389/fphys.2019.01531. eCollection 2019. Front Physiol. 2020. PMID: 31969828 Free PMC article. Review.
-
Targeting AMPK Signaling as a Neuroprotective Strategy in Parkinson's Disease.J Parkinsons Dis. 2018;8(2):161-181. doi: 10.3233/JPD-171296. J Parkinsons Dis. 2018. PMID: 29614701 Free PMC article. Review.
-
Sex-specific decline in prefrontal cortex mitochondrial bioenergetics in aging baboons correlates with walking speed.bioRxiv [Preprint]. 2024 Sep 24:2024.09.19.613684. doi: 10.1101/2024.09.19.613684. bioRxiv. 2024. Update in: Neurobiol Aging. 2025 Jul;151:1-12. doi: 10.1016/j.neurobiolaging.2025.03.010. PMID: 39386547 Free PMC article. Updated. Preprint.
References
-
- Aguirre V, Uchida T, Yenush L, Davis R, and White MF. The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307). J Biol Chem 275: 9047–9054, 2000 - PubMed
-
- Ahn JH, Choi JH, Song JM, Lee CH, Yoo KY, Hwang IK, Kim JS, Shin HC, and Won MH. Increase in Trx2/Prx3 redox system immunoreactivity in the spinal cord and hippocampus of aged dogs. Exp Gerontol 46: 946–952, 2011 - PubMed
-
- Al-Regaiey KA, Masternak MM, Bonkowski M, Sun L, and Bartke A. Long-lived growth hormone receptor knockout mice: interaction of reduced insulin-like growth factor i/insulin signaling and caloric restriction. Endocrinology 146: 851–860, 2005 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
