

Biological and therapeutic relevance of nonreplicative DNA polymerases to cancer
- PMID: 22794079
- PMCID: PMC3557440
- DOI: 10.1089/ars.2011.4203
Biological and therapeutic relevance of nonreplicative DNA polymerases to cancer
Abstract
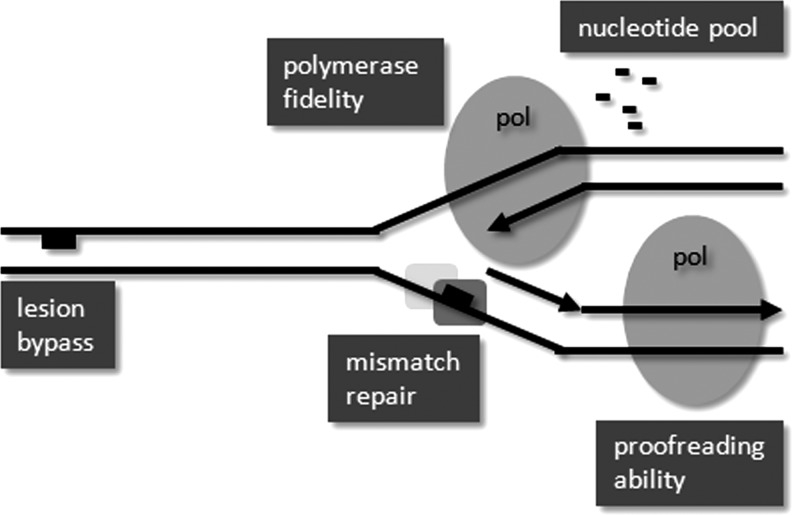
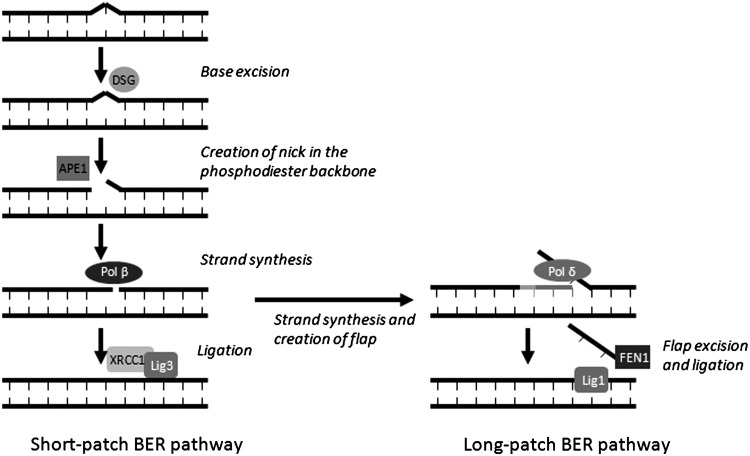
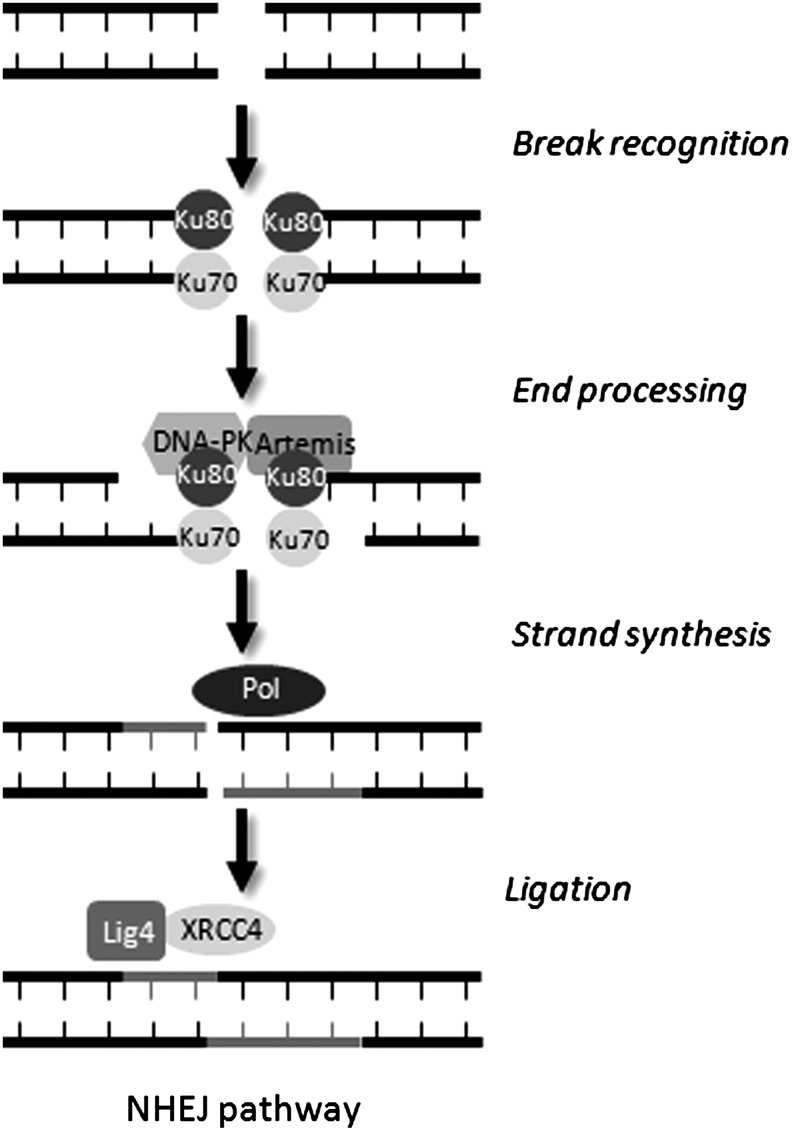
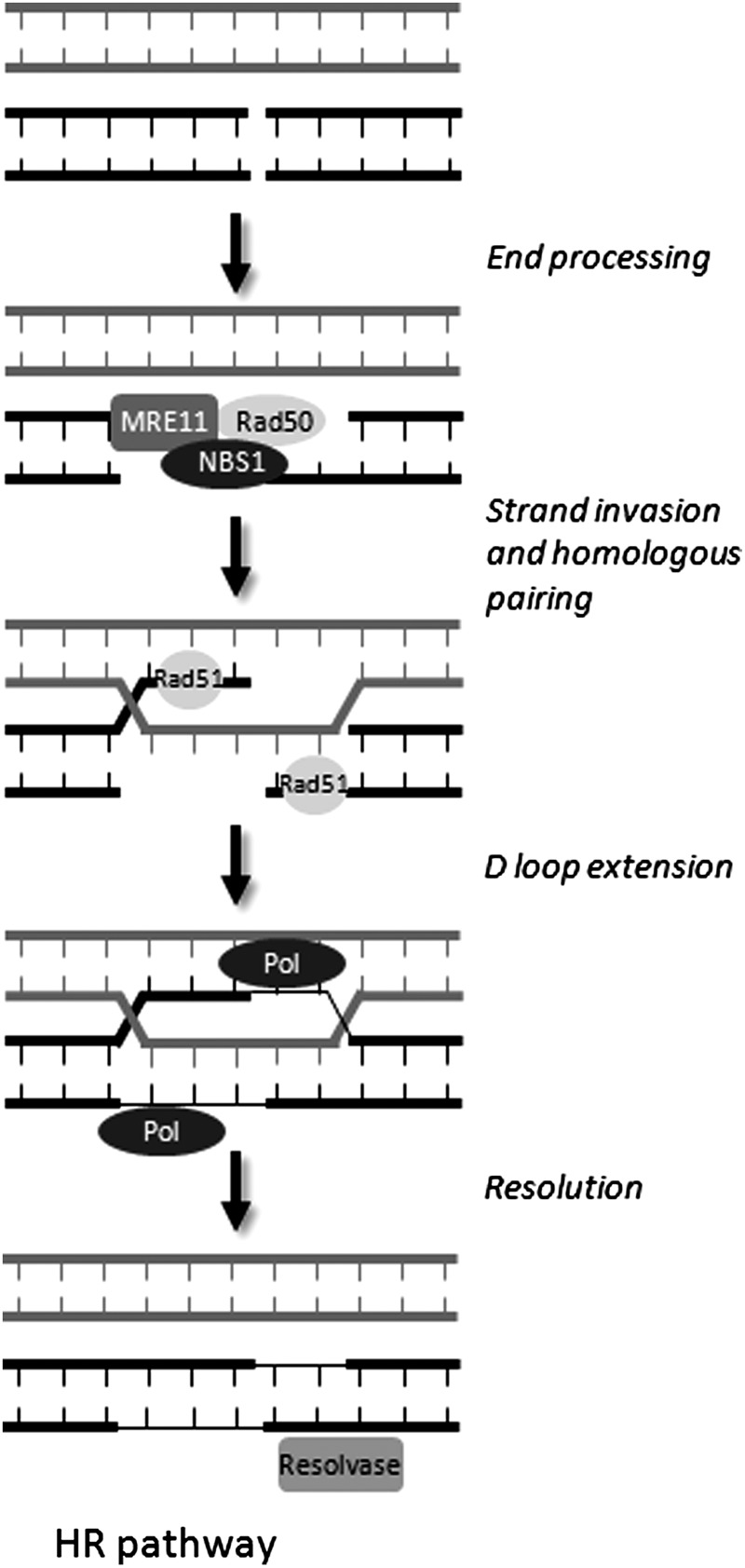
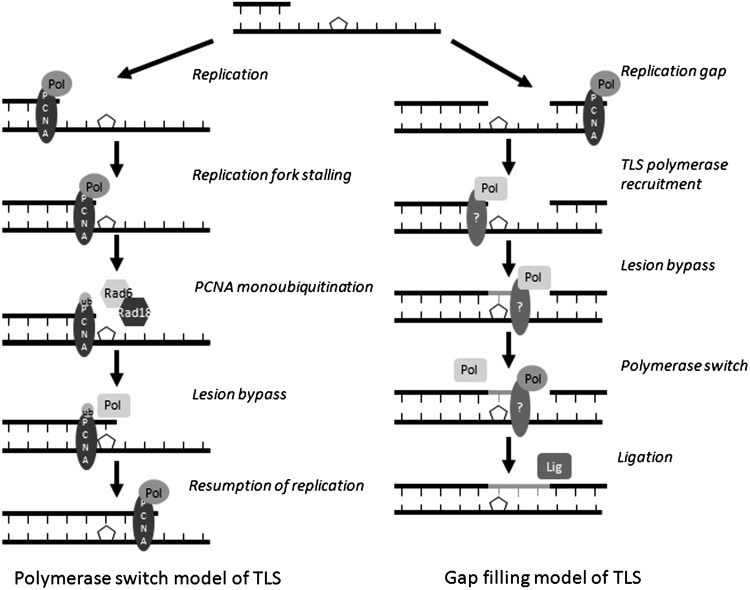
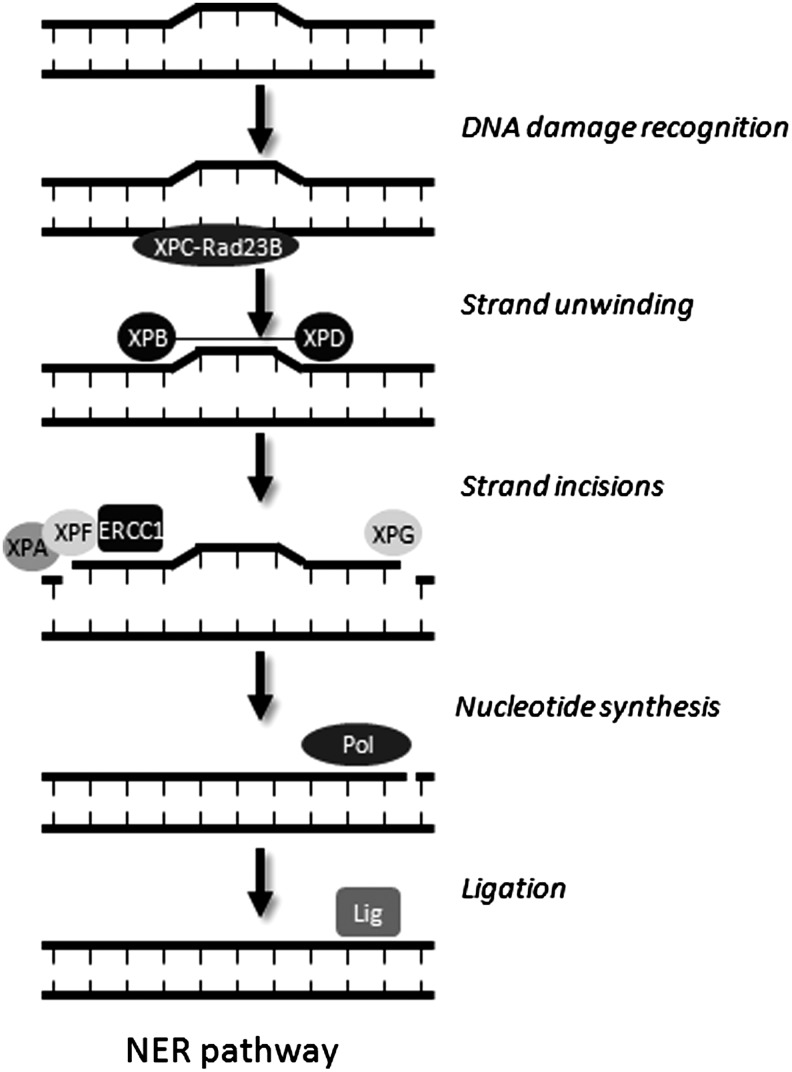
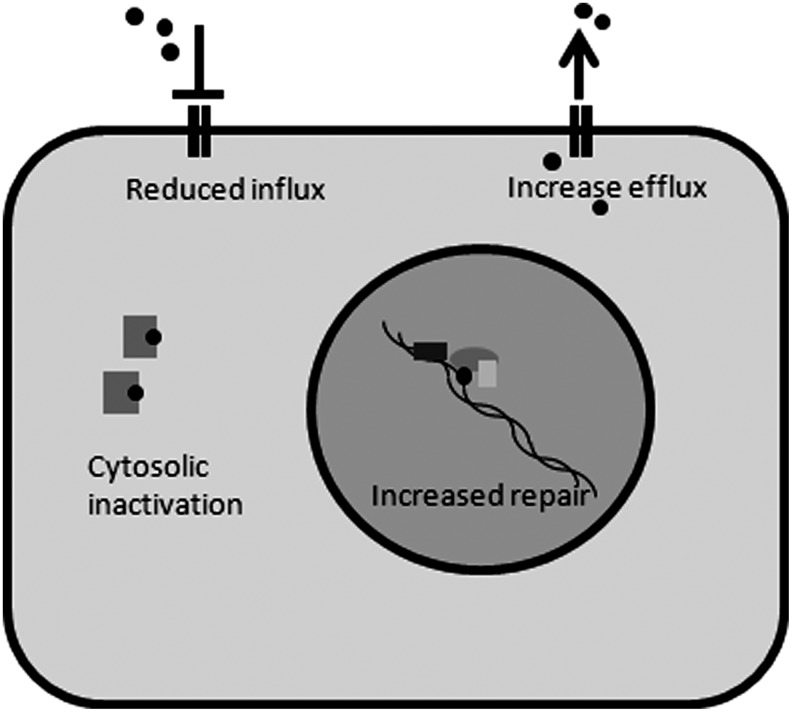
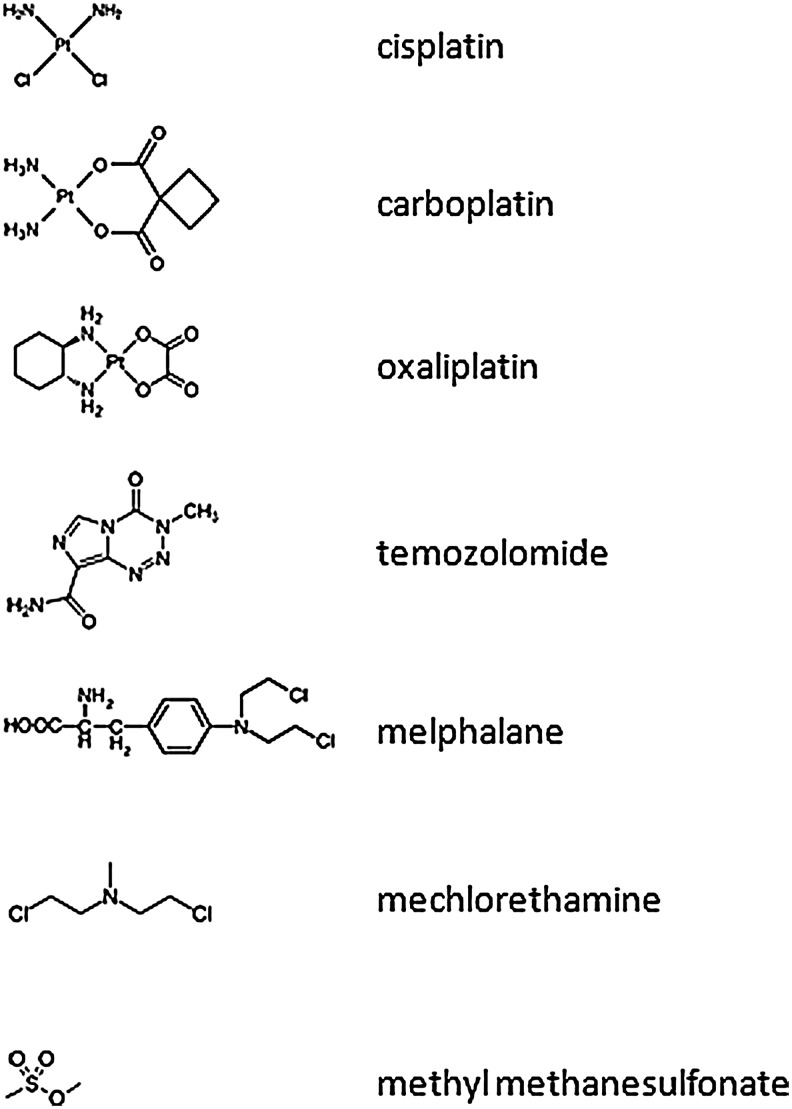
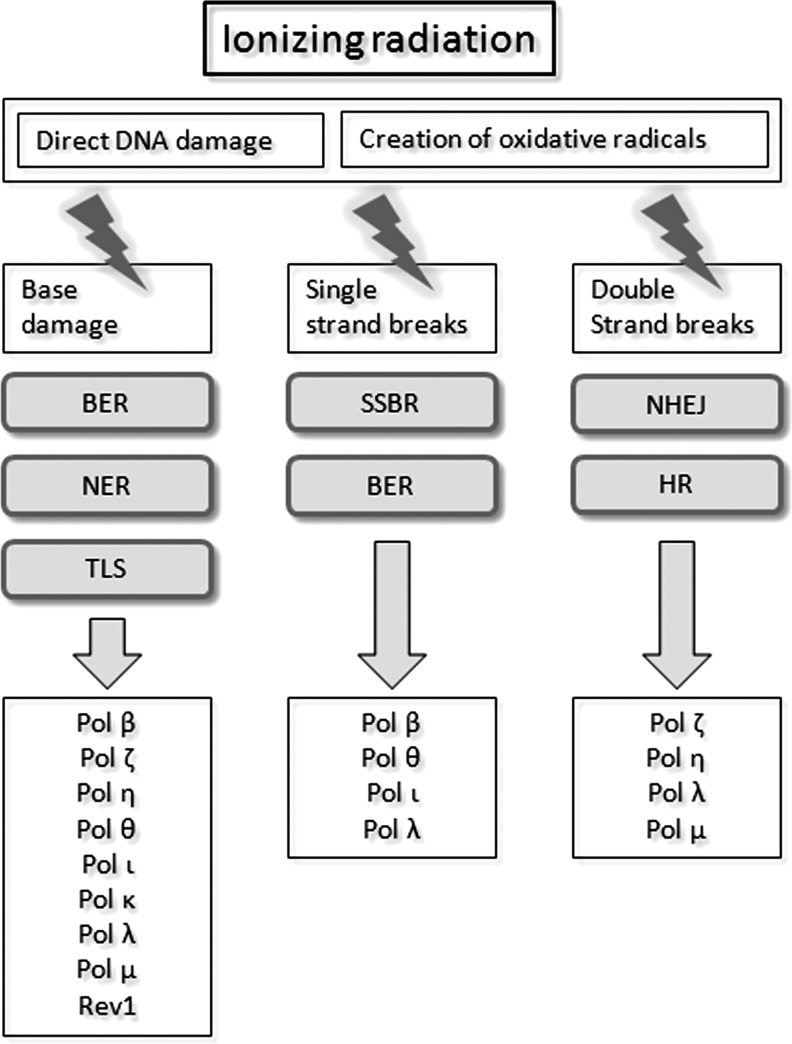
Apart from surgical approaches, the treatment of cancer remains largely underpinned by radiotherapy and pharmacological agents that cause damage to cellular DNA, which ultimately causes cancer cell death. DNA polymerases, which are involved in the repair of cellular DNA damage, are therefore potential targets for inhibitors for improving the efficacy of cancer therapy. They can be divided, according to their main function, into two groups, namely replicative and nonreplicative enzymes. At least 15 different DNA polymerases, including their homologs, have been discovered to date, which vary considerably in processivity and fidelity. Many of the nonreplicative (specialized) DNA polymerases replicate DNA in an error-prone fashion, and they have been shown to participate in multiple DNA damage repair and tolerance pathways, which are often aberrant in cancer cells. Alterations in DNA repair pathways involving DNA polymerases have been linked with cancer survival and with treatment response to radiotherapy or to classes of cytotoxic drugs routinely used for cancer treatment, particularly cisplatin, oxaliplatin, etoposide, and bleomycin. Indeed, there are extensive preclinical data to suggest that DNA polymerase inhibition may prove to be a useful approach for increasing the effectiveness of therapies in patients with cancer. Furthermore, specialized DNA polymerases warrant examination of their potential use as clinical biomarkers to select for particular cancer therapies, to individualize treatment for patients.
Figures
References
-
- Adachi M. Ijichi K. Hasegawa Y. Ogawa T. Nakamura H. Yasui Y. Fukushima M. Ishizaki K. Hypersensitivity to cisplatin after hRev3 mRNA knockdown in head and neck squamous cell carcinoma cells. Mol Med Report. 2008;1:695–698. - PubMed
-
- Albertella MR. Green CM. Lehmann AR. O'Connor MJ. A role for polymerase eta in the cellular tolerance to cisplatin-induced damage. Cancer Res. 2005;65:9799–9806. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
