

IL-17A production by renal γδ T cells promotes kidney injury in crescentic GN
- PMID: 22797181
- PMCID: PMC3431412
- DOI: 10.1681/ASN.2012010040
IL-17A production by renal γδ T cells promotes kidney injury in crescentic GN
Abstract
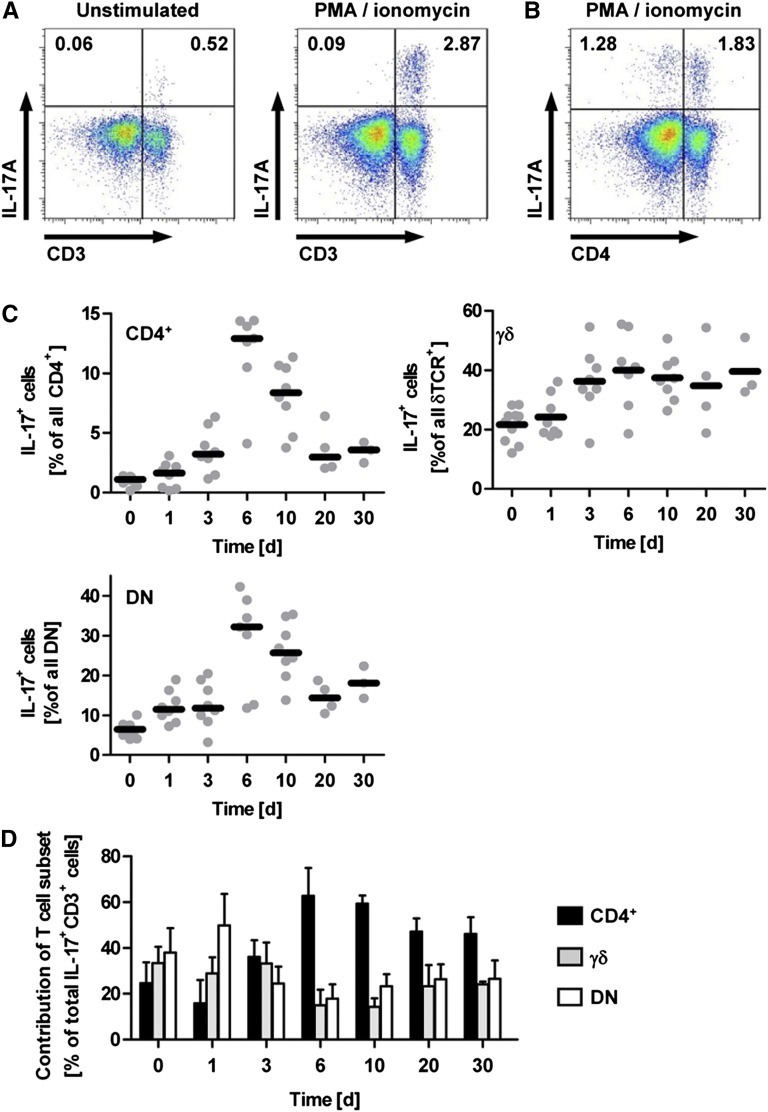
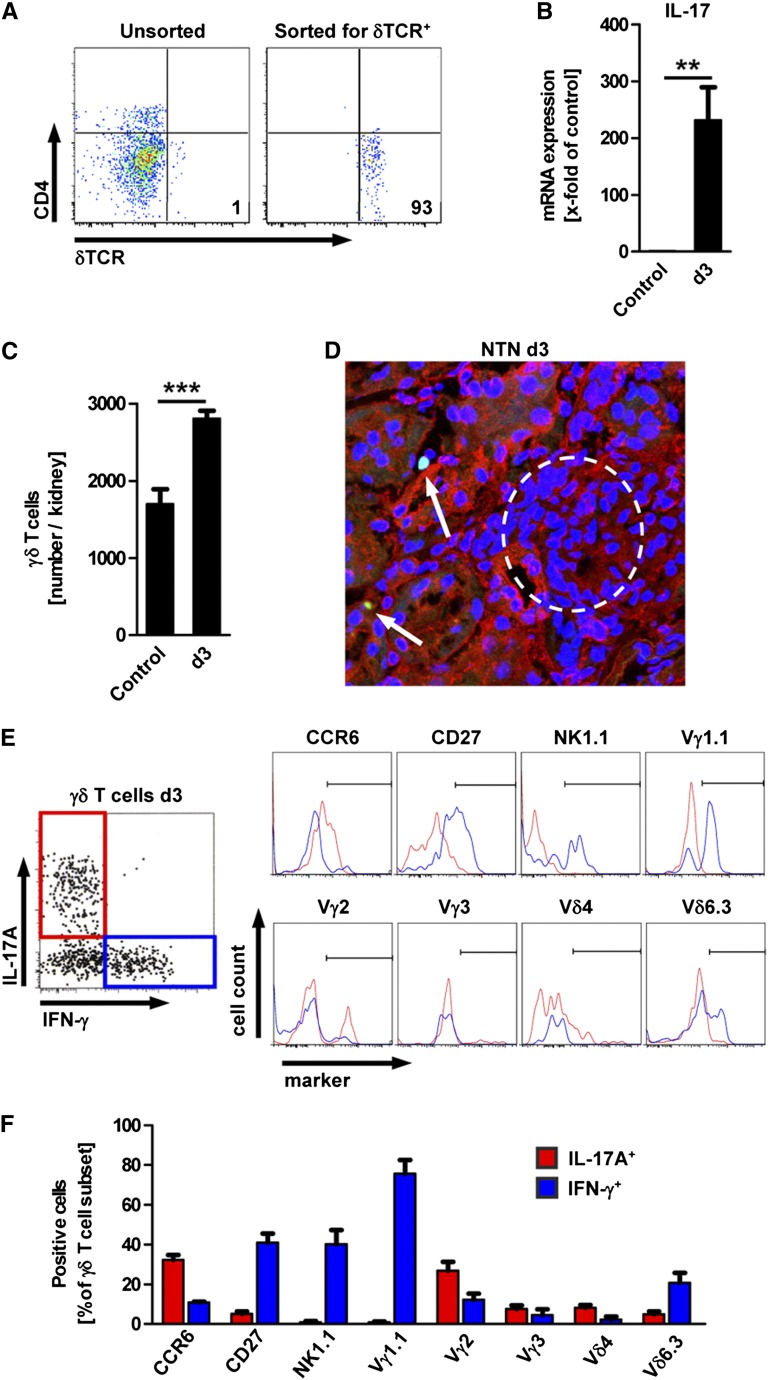
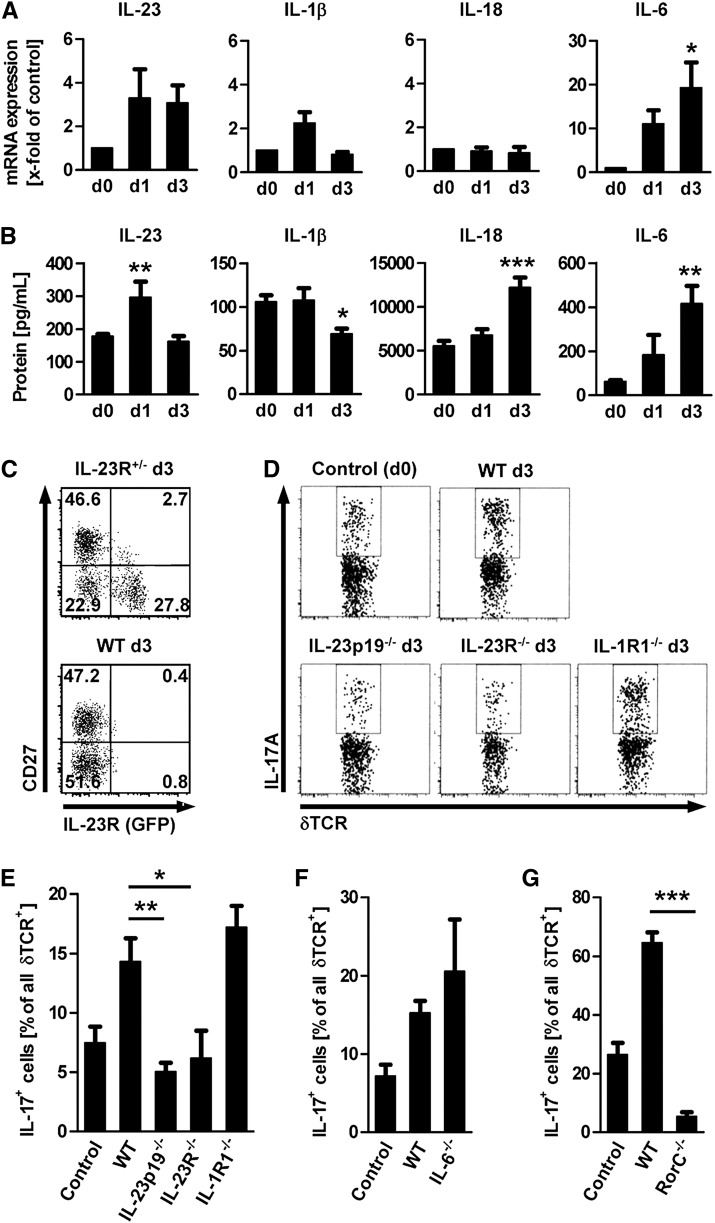
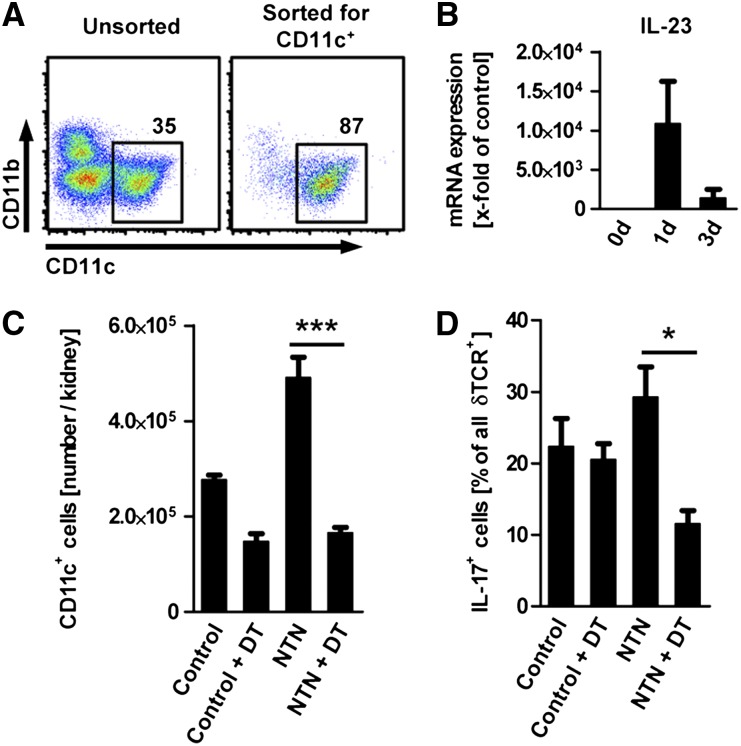
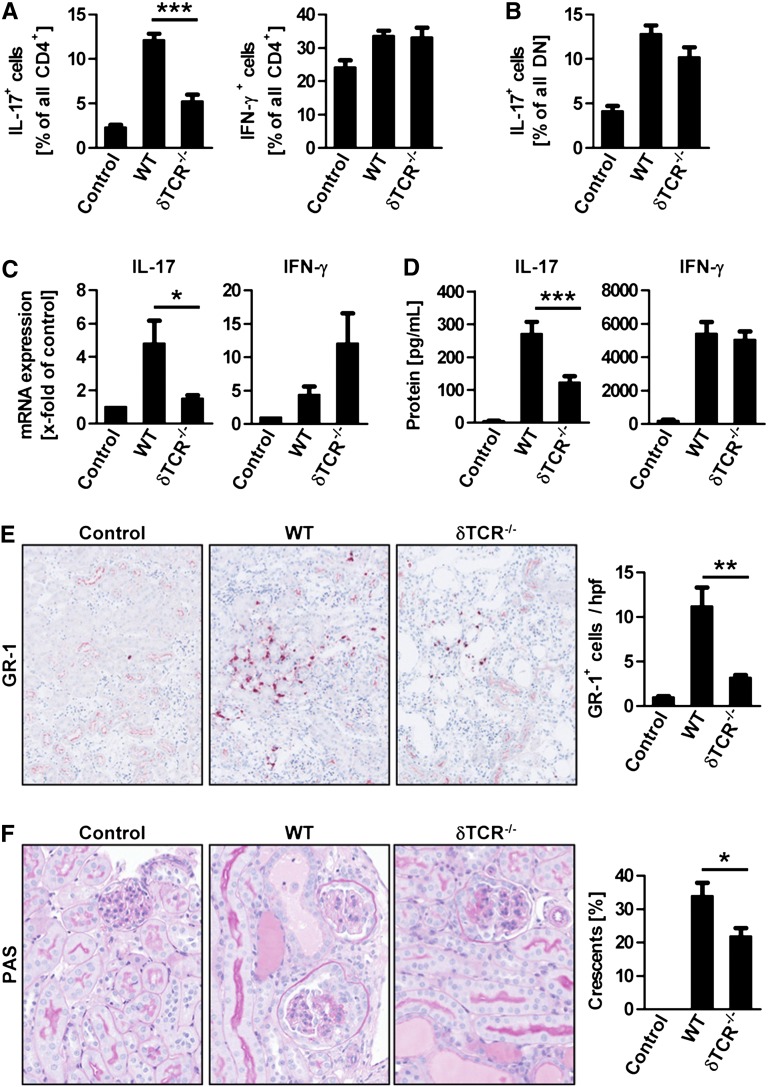
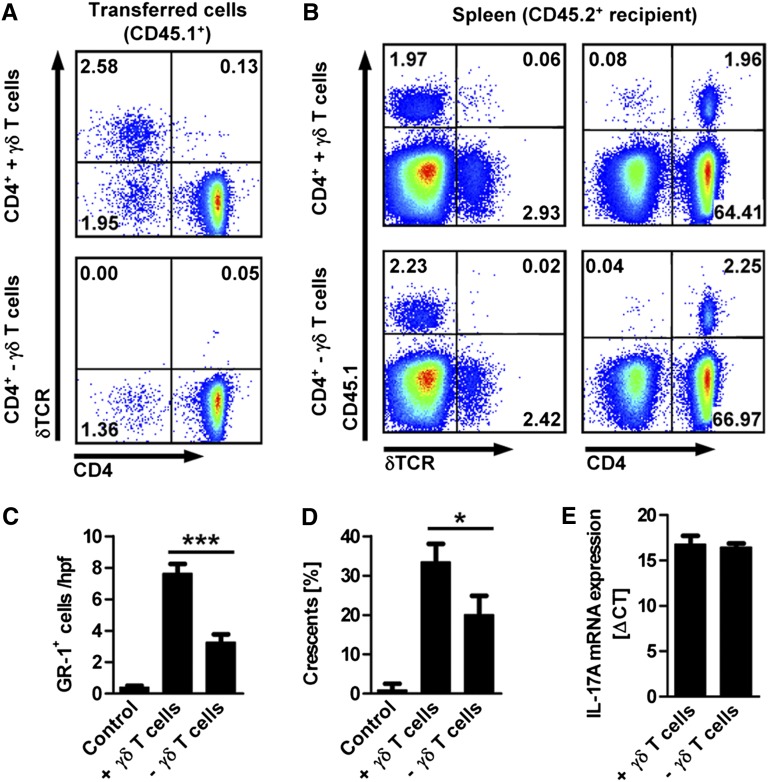
The Th17 immune response appears to contribute to the pathogenesis of human and experimental crescentic GN, but the cell types that produce IL-17A in the kidney, the mechanisms involved in its induction, and the IL-17A-mediated effector functions that promote renal tissue injury are incompletely understood. Here, using a murine model of crescentic GN, we found that CD4(+) T cells, γδ T cells, and a population of CD3(+)CD4(-)CD8(-)γδT cell receptor(-)NK1.1(-) T cells all produce IL-17A in the kidney. A time course analysis identified γδ T cells as a major source of IL-17A in the early phase of disease, before the first CD4(+) Th17 cells arrived. The production of IL-17A by renal γδ T cells depended on IL-23p19 signaling and retinoic acid-related orphan receptor-γt but not on IL-1β or IL-6. In addition, depletion of dendritic cells, which produce IL-23 in the kidney, reduced IL-17A production by renal γδ T cells. Furthermore, the lack of IL-17A production in γδ T cells, as well as the absence of all γδ T cells, reduced neutrophil recruitment into the kidney and ameliorated renal injury. Taken together, these data suggest that γδ T cells produce IL-17A in the kidney, induced by IL-23, promoting neutrophil recruitment, and contributing to the immunopathogenesis of crescentic GN.
Figures
Similar articles
-
Interleukin-23 is critical for full-blown expression of a non-autoimmune destructive arthritis and regulates interleukin-17A and RORgammat in gammadelta T cells.Arthritis Res Ther. 2009;11(6):R194. doi: 10.1186/ar2893. Epub 2009 Dec 17. Arthritis Res Ther. 2009. PMID: 20017902 Free PMC article.
-
IL-17C/IL-17 Receptor E Signaling in CD4+ T Cells Promotes TH17 Cell-Driven Glomerular Inflammation.J Am Soc Nephrol. 2018 Apr;29(4):1210-1222. doi: 10.1681/ASN.2017090949. Epub 2018 Feb 26. J Am Soc Nephrol. 2018. PMID: 29483158 Free PMC article.
-
Pathogenic Role for γδ T Cells in Autoimmune Anti-Myeloperoxidase Glomerulonephritis.J Immunol. 2017 Nov 1;199(9):3042-3050. doi: 10.4049/jimmunol.1602025. Epub 2017 Sep 27. J Immunol. 2017. PMID: 28954887
-
Regulation and function of IL-17A- and IL-22-producing γδ T cells.Cell Mol Life Sci. 2011 Jul;68(14):2371-90. doi: 10.1007/s00018-011-0700-z. Epub 2011 May 15. Cell Mol Life Sci. 2011. PMID: 21573786 Free PMC article. Review.
-
Whodunit? The Contribution of Interleukin (IL)-17/IL-22-Producing γδ T Cells, αβ T Cells, and Innate Lymphoid Cells to the Pathogenesis of Spondyloarthritis.Front Immunol. 2018 Apr 25;9:885. doi: 10.3389/fimmu.2018.00885. eCollection 2018. Front Immunol. 2018. PMID: 29922283 Free PMC article. Review.
Cited by
-
The immune system and kidney disease: basic concepts and clinical implications.Nat Rev Immunol. 2013 Oct;13(10):738-53. doi: 10.1038/nri3523. Epub 2013 Sep 16. Nat Rev Immunol. 2013. PMID: 24037418 Review.
-
Myocardial infarction worsens glomerular injury and microalbuminuria in rats with pre-existing renal impairment accompanied by the activation of ER stress and inflammation.Mol Biol Rep. 2014 Dec;41(12):7911-21. doi: 10.1007/s11033-014-3685-5. Epub 2014 Aug 31. Mol Biol Rep. 2014. PMID: 25173645
-
Deletion of HIF-2α in Dendritic Cells Attenuates Anti-Glomerular Basement Membrane Nephritis.Biomedicines. 2025 Apr 7;13(4):888. doi: 10.3390/biomedicines13040888. Biomedicines. 2025. PMID: 40299471 Free PMC article.
-
RTEC-intrinsic IL-17-driven inflammatory circuit amplifies antibody-induced glomerulonephritis and is constrained by Regnase-1.JCI Insight. 2021 Jul 8;6(13):e147505. doi: 10.1172/jci.insight.147505. JCI Insight. 2021. PMID: 34236049 Free PMC article.
-
IL-17 in Renal Immunity and Autoimmunity.J Immunol. 2018 Dec 1;201(11):3153-3159. doi: 10.4049/jimmunol.1801042. J Immunol. 2018. PMID: 30455371 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
