

Architectural and thermodynamic principles underlying intramembrane protease function
- PMID: 22797666
- PMCID: PMC4028635
- DOI: 10.1038/nchembio.1021
Architectural and thermodynamic principles underlying intramembrane protease function
Abstract
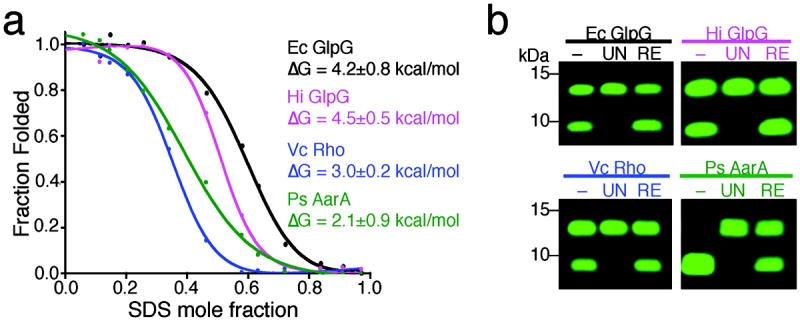
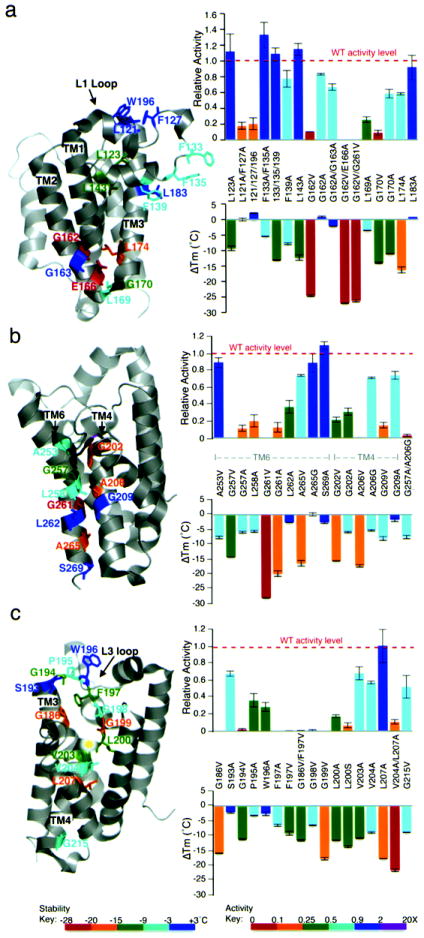
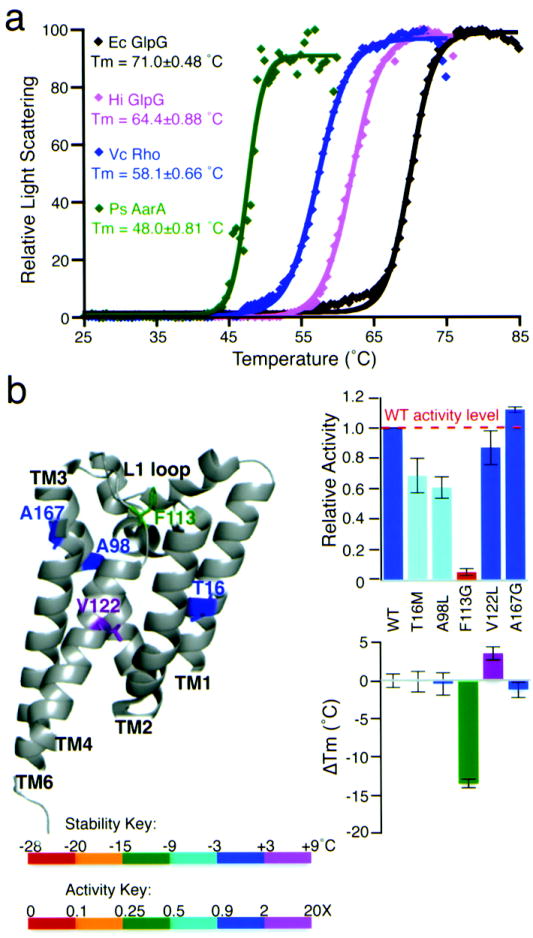
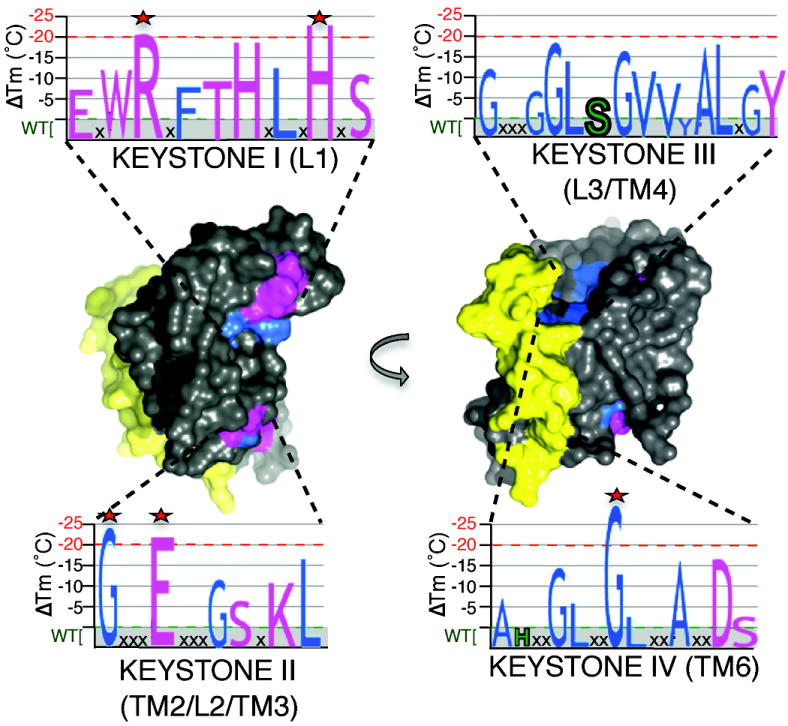
Intramembrane proteases hydrolyze peptide bonds within the membrane as a signaling paradigm universal to all life forms and with implications in disease. Deciphering the architectural strategies supporting intramembrane proteolysis is an essential but unattained goal. We integrated new, quantitative and high-throughput thermal light-scattering technology, reversible equilibrium unfolding and refolding and quantitative protease assays to interrogate rhomboid architecture with 151 purified variants. Rhomboid proteases maintain low intrinsic thermodynamic stability (ΔG = 2.1-4.5 kcal mol(-1)) resulting from a multitude of generally weak transmembrane packing interactions, making them highly responsive to their environment. Stability is consolidated by two buried glycines and several packing leucines, with a few multifaceted hydrogen bonds strategically deployed to two peripheral regions. Opposite these regions lie transmembrane segment 5 and connected loops that are notably exempt of structural responsibility, suggesting intramembrane proteolysis involves considerable but localized protein dynamics. Our analyses provide a comprehensive 'heat map' of the physiochemical anatomy underlying membrane-immersed enzyme function at, what is to our knowledge, unprecedented resolution.
Conflict of interest statement
The authors declare no conflict of interest exists.
Figures




Similar articles
-
Enzymatic analysis of a rhomboid intramembrane protease implicates transmembrane helix 5 as the lateral substrate gate.Proc Natl Acad Sci U S A. 2007 May 15;104(20):8257-62. doi: 10.1073/pnas.0700814104. Epub 2007 Apr 26. Proc Natl Acad Sci U S A. 2007. PMID: 17463085 Free PMC article.
-
Reconstitution of intramembrane proteolysis in vitro reveals that pure rhomboid is sufficient for catalysis and specificity.Proc Natl Acad Sci U S A. 2005 Feb 8;102(6):1883-8. doi: 10.1073/pnas.0408306102. Epub 2005 Jan 31. Proc Natl Acad Sci U S A. 2005. PMID: 15684070 Free PMC article.
-
Sensitive Versatile Fluorogenic Transmembrane Peptide Substrates for Rhomboid Intramembrane Proteases.J Biol Chem. 2017 Feb 17;292(7):2703-2713. doi: 10.1074/jbc.M116.762849. Epub 2017 Jan 9. J Biol Chem. 2017. PMID: 28069810 Free PMC article.
-
Core principles of intramembrane proteolysis: comparison of rhomboid and site-2 family proteases.Curr Opin Struct Biol. 2008 Aug;18(4):432-41. doi: 10.1016/j.sbi.2008.03.005. Epub 2008 Apr 26. Curr Opin Struct Biol. 2008. PMID: 18440799 Free PMC article. Review.
-
The discovery of proteases and intramembrane proteolysis 1.Biochem Cell Biol. 2019 Jun;97(3):265-269. doi: 10.1139/bcb-2018-0186. Epub 2018 Aug 13. Biochem Cell Biol. 2019. PMID: 30102867 Review.
Cited by
-
Decoding the Functional Evolution of an Intramembrane Protease Superfamily by Statistical Coupling Analysis.Structure. 2020 Dec 1;28(12):1329-1336.e4. doi: 10.1016/j.str.2020.07.015. Epub 2020 Aug 13. Structure. 2020. PMID: 32795403 Free PMC article.
-
Factors That Control the Force Needed to Unfold a Membrane Protein in Silico Depend on the Mode of Denaturation.Int J Mol Sci. 2023 Jan 31;24(3):2654. doi: 10.3390/ijms24032654. Int J Mol Sci. 2023. PMID: 36768981 Free PMC article.
-
Energy landscape underlying spontaneous insertion and folding of an alpha-helical transmembrane protein into a bilayer.Nat Commun. 2018 Nov 23;9(1):4949. doi: 10.1038/s41467-018-07320-9. Nat Commun. 2018. PMID: 30470737 Free PMC article.
-
Rhomboid intramembrane protease YqgP licenses bacterial membrane protein quality control as adaptor of FtsH AAA protease.EMBO J. 2020 May 18;39(10):e102935. doi: 10.15252/embj.2019102935. Epub 2020 Jan 13. EMBO J. 2020. PMID: 31930742 Free PMC article.
-
Design of self-assembling transmembrane helical bundles to elucidate principles required for membrane protein folding and ion transport.Philos Trans R Soc Lond B Biol Sci. 2017 Aug 5;372(1726):20160214. doi: 10.1098/rstb.2016.0214. Philos Trans R Soc Lond B Biol Sci. 2017. PMID: 28630154 Free PMC article. Review.
References
-
- Bowie JU. Solving the membrane protein folding problem. Nature. 2005;438:581–9. - PubMed
-
- De Strooper B, Annaert W. Novel research horizons for presenilins and gamma-secretases in cell biology and disease. Annu Rev Cell Dev Biol. 2010;26:235–60. - PubMed
-
- Brown MS, Ye J, Rawson RB, Goldstein JL. Regulated intramembrane proteolysis: a control mechanism conserved from bacteria to humans. Cell. 2000;100:391–8. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
