

Fibrillation precursor of superoxide dismutase 1 revealed by gradual tuning of the protein-folding equilibrium
- PMID: 22797895
- PMCID: PMC3497812
- DOI: 10.1073/pnas.1201795109
Fibrillation precursor of superoxide dismutase 1 revealed by gradual tuning of the protein-folding equilibrium
Abstract
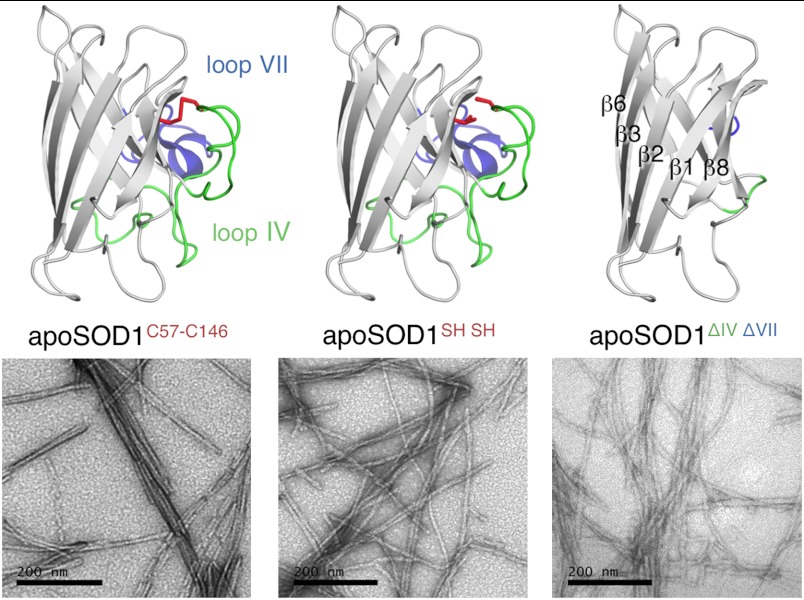
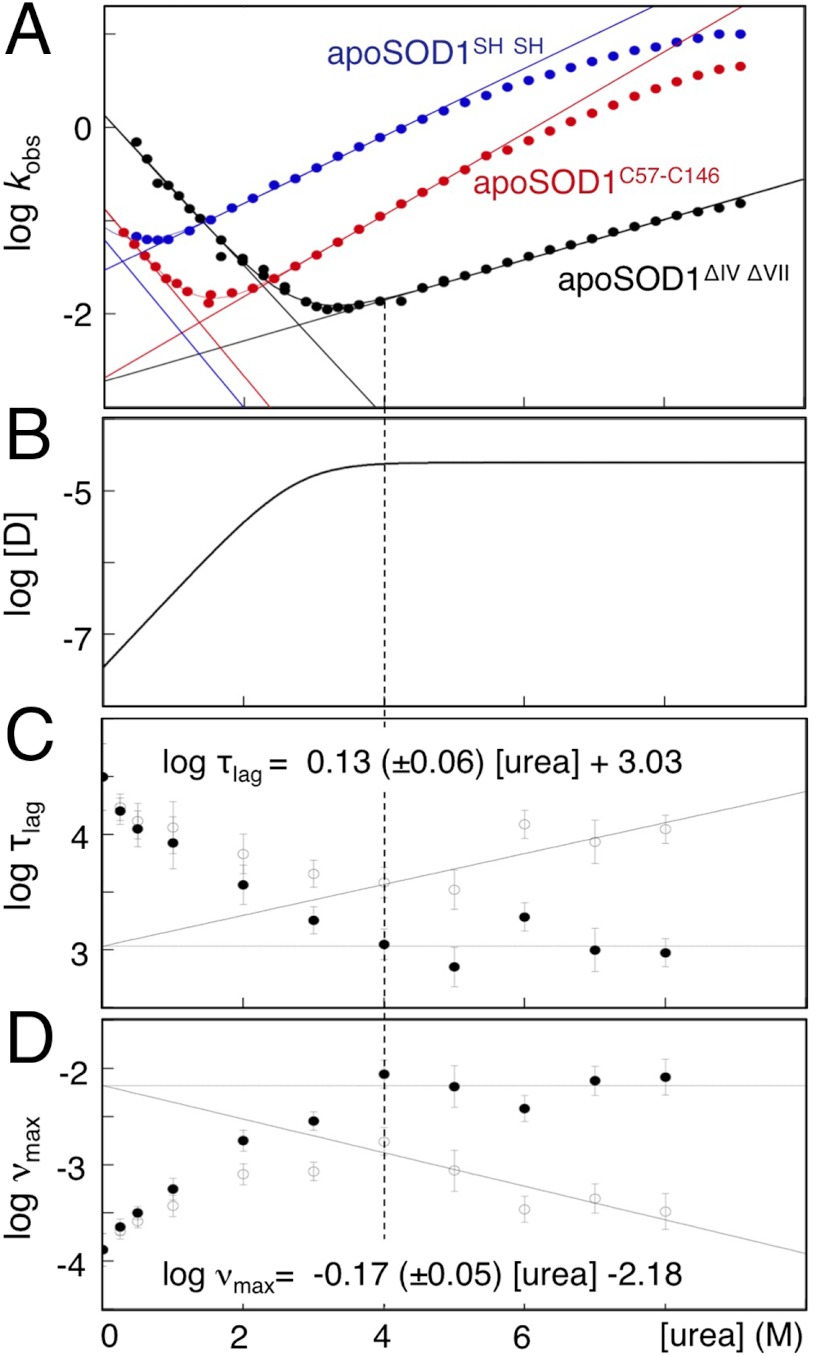
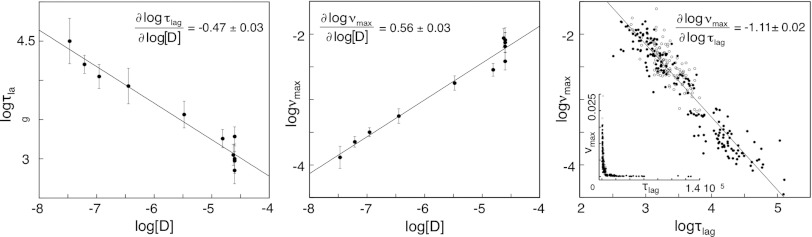
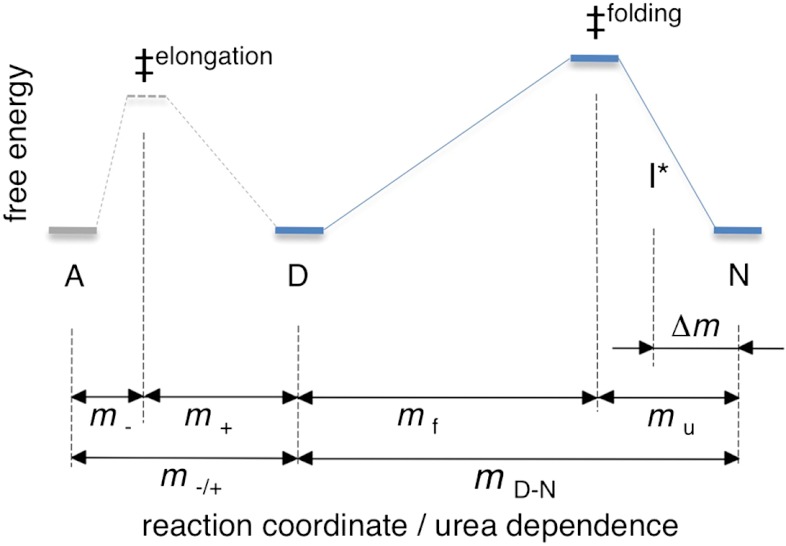
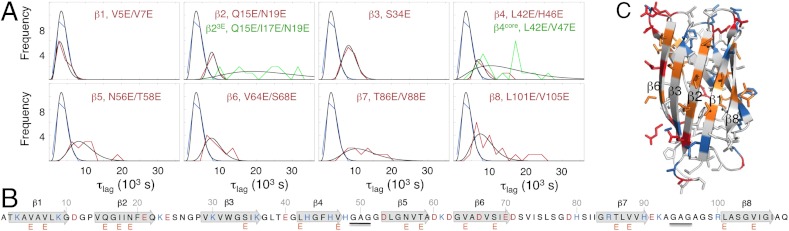
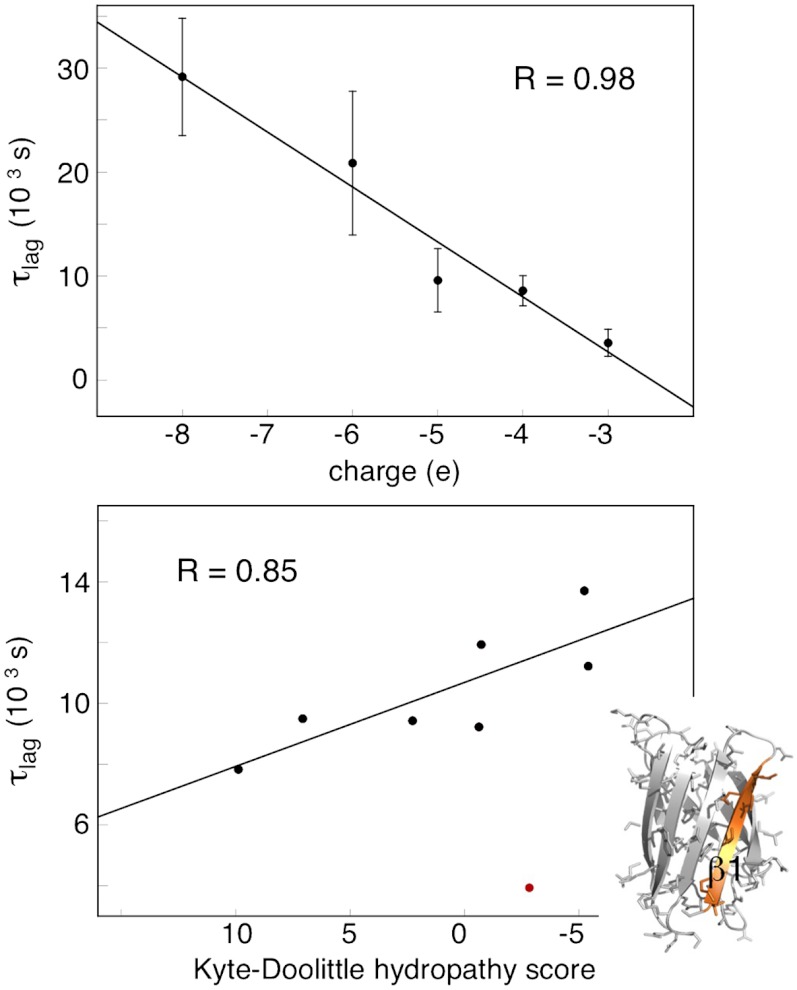
Although superoxide dismutase 1 (SOD1) stands out as a relatively soluble protein in vitro, it can be made to fibrillate by mechanical agitation. The mechanism of this fibrillation process is yet poorly understood, but attains considerable interest due to SOD1's involvement in the neurodegenerative disease amyotrophic lateral sclerosis (ALS). In this study, we map out the apoSOD1 fibrillation process from how it competes with the global folding events at increasing concentrations of urea: We determine how the fibrillation lag time (τ(lag)) and maximum growth rate (ν(max)) depend on gradual titration of the folding equilibrium, from the native to the unfolded state. The results show that the agitation-induced fibrillation of apoSOD1 uses globally unfolded precursors and relies on fragmentation-assisted growth. Mutational screening and fibrillation m-values (∂ log τ(lag)/∂[urea] and ∂ log ν(max)/∂[urea]) indicate moreover that the fibrillation pathway proceeds via a diffusely bound transient complex that responds to the global physiochemical properties of the SOD1 sequence. Fibrillation of apoSOD1, as it bifurcates from the denatured ensemble, seems thus mechanistically analogous to that of disordered peptides, save the competing folding transition to the native state. Finally, we examine by comparison with in vivo data to what extent this mode of fibrillation, originating from selective amplification of mechanically brittle aggregates by sample agitation, captures the mechanism of pathological SOD1 aggregation in ALS.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Nonnative structure in a peptide model of the unfolded state of superoxide dismutase 1 (SOD1): Implications for ALS-linked aggregation.J Biol Chem. 2019 Sep 13;294(37):13708-13717. doi: 10.1074/jbc.RA119.008765. Epub 2019 Jul 24. J Biol Chem. 2019. PMID: 31341015 Free PMC article.
-
Thermal fluctuations of immature SOD1 lead to separate folding and misfolding pathways.Elife. 2015 Jun 23;4:e07296. doi: 10.7554/eLife.07296. Elife. 2015. PMID: 26099300 Free PMC article.
-
Enthalpic barriers dominate the folding and unfolding of the human Cu, Zn superoxide dismutase monomer.J Mol Biol. 2012 Dec 7;424(3-4):192-202. doi: 10.1016/j.jmb.2012.09.009. Epub 2012 Sep 18. J Mol Biol. 2012. PMID: 22999954 Free PMC article.
-
SOD1 aggregation and ALS: role of metallation states and disulfide status.Curr Top Med Chem. 2012;12(22):2560-72. doi: 10.2174/1568026611212220010. Curr Top Med Chem. 2012. PMID: 23339308 Review.
-
Does wild-type Cu/Zn-superoxide dismutase have pathogenic roles in amyotrophic lateral sclerosis?Transl Neurodegener. 2020 Aug 19;9(1):33. doi: 10.1186/s40035-020-00209-y. Transl Neurodegener. 2020. PMID: 32811540 Free PMC article. Review.
Cited by
-
Understanding protein aggregation from the view of monomer dynamics.Mol Biosyst. 2013 Jan 27;9(1):29-35. doi: 10.1039/c2mb25334h. Epub 2012 Oct 26. Mol Biosyst. 2013. PMID: 23104145 Free PMC article. Review.
-
N-terminal acetylation of superoxide dismutase 1 accelerates amyloid formation without general destabilization of the apo state.Protein Sci. 2025 Sep;34(9):e70267. doi: 10.1002/pro.70267. Protein Sci. 2025. PMID: 40815260 Free PMC article.
-
Mutant SOD1 aggregates formed in vitro and in cultured cells are polymorphic and differ from those arising in the CNS.J Neurochem. 2023 Jan;164(1):77-93. doi: 10.1111/jnc.15718. Epub 2022 Nov 23. J Neurochem. 2023. PMID: 36326589 Free PMC article.
-
Amyotrophic Lateral Sclerosis: Proteins, Proteostasis, Prions, and Promises.Front Cell Neurosci. 2020 Nov 4;14:581907. doi: 10.3389/fncel.2020.581907. eCollection 2020. Front Cell Neurosci. 2020. PMID: 33328890 Free PMC article.
-
Proteostasis and Its Role in Disease Development.Cell Biochem Biophys. 2025 Jun;83(2):1725-1741. doi: 10.1007/s12013-024-01581-6. Epub 2024 Oct 18. Cell Biochem Biophys. 2025. PMID: 39422790 Free PMC article. Review.
References
-
- Eichner T, Radford SE. A diversity of assembly mechanisms of a generic amyloid fold. Mol Cell. 2011;43:8–18. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
