

Congenital myopathy-causing tropomyosin mutations induce thin filament dysfunction via distinct physiological mechanisms
- PMID: 22798622
- PMCID: PMC3459469
- DOI: 10.1093/hmg/dds289
Congenital myopathy-causing tropomyosin mutations induce thin filament dysfunction via distinct physiological mechanisms
Abstract
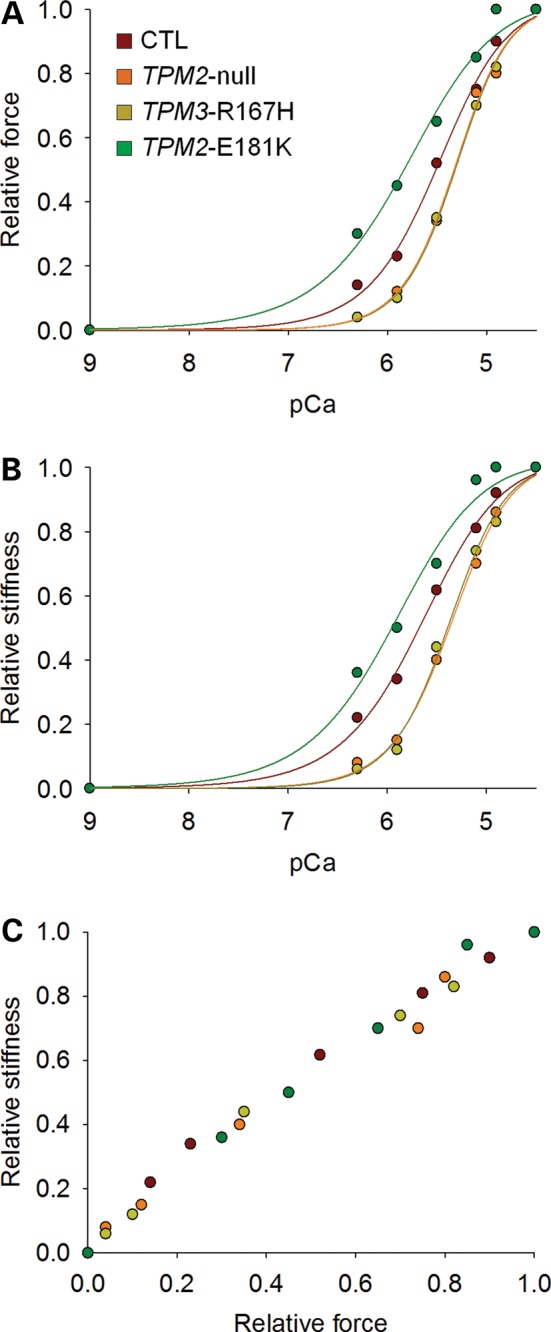
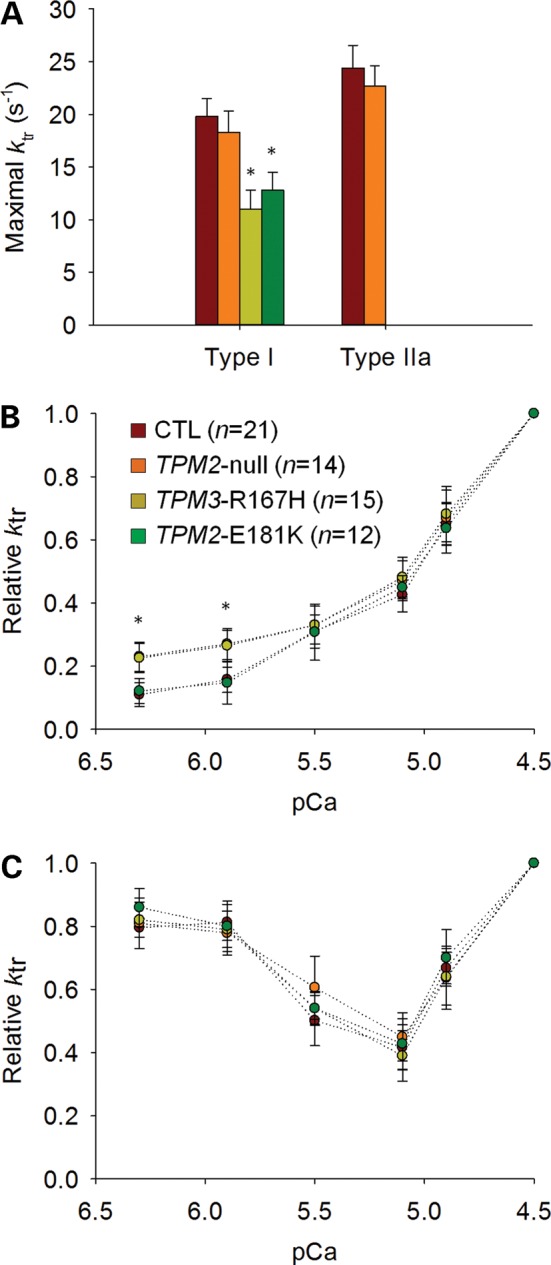
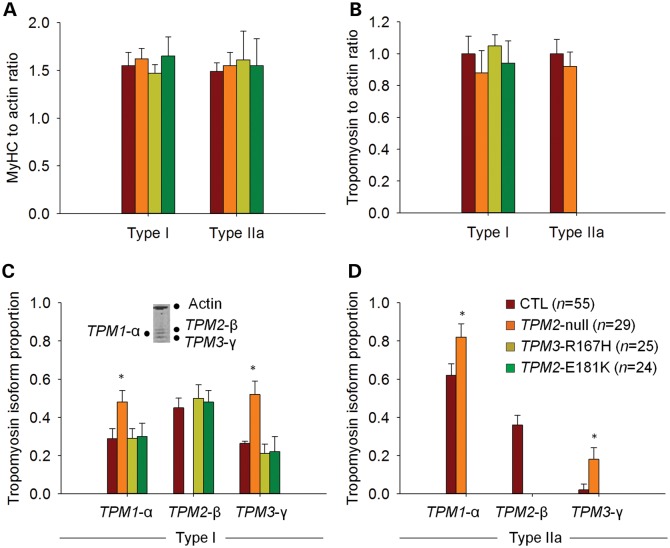
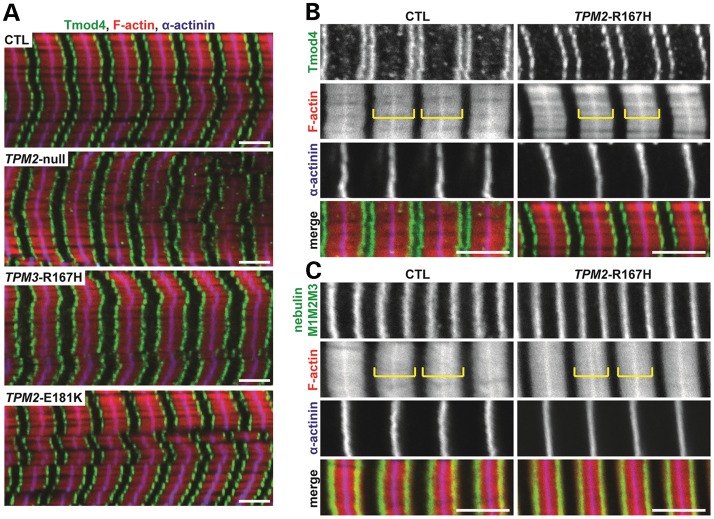
In humans, congenital myopathy-linked tropomyosin mutations lead to skeletal muscle dysfunction, but the cellular and molecular mechanisms underlying such dysfunction remain obscure. Recent studies have suggested a unifying mechanism by which tropomyosin mutations partially inhibit thin filament activation and prevent proper formation and cycling of myosin cross-bridges, inducing force deficits at the fiber and whole-muscle levels. Here, we aimed to verify this mechanism using single membrane-permeabilized fibers from patients with three tropomyosin mutations (TPM2-null, TPM3-R167H and TPM2-E181K) and measuring a broad range of parameters. Interestingly, we identified two divergent, mutation-specific pathophysiological mechanisms. (i) The TPM2-null and TPM3-R167H mutations both decreased cooperative thin filament activation in combination with reductions in the myosin cross-bridge number and force production. The TPM3-R167H mutation also induced a concomitant reduction in thin filament length. (ii) In contrast, the TPM2-E181K mutation increased thin filament activation, cross-bridge binding and force generation. In the former mechanism, modulating thin filament activation by administering troponin activators (CK-1909178 and EMD 57033) to single membrane-permeabilized fibers carrying tropomyosin mutations rescued the thin filament activation defect associated with the pathophysiology. Therefore, administration of troponin activators may constitute a promising therapeutic approach in the future.
Figures
Similar articles
-
Thin filament dysfunctions caused by mutations in tropomyosin Tpm3.12 and Tpm1.1.J Muscle Res Cell Motil. 2020 Mar;41(1):39-53. doi: 10.1007/s10974-019-09532-y. Epub 2019 Jul 3. J Muscle Res Cell Motil. 2020. PMID: 31270709 Free PMC article. Review.
-
Mutation update and genotype-phenotype correlations of novel and previously described mutations in TPM2 and TPM3 causing congenital myopathies.Hum Mutat. 2014 Jul;35(7):779-90. doi: 10.1002/humu.22554. Epub 2014 May 1. Hum Mutat. 2014. PMID: 24692096 Free PMC article.
-
Congenital myopathy-related mutations in tropomyosin disrupt regulatory function through altered actin affinity and tropomodulin binding.FEBS J. 2019 May;286(10):1877-1893. doi: 10.1111/febs.14787. Epub 2019 Mar 5. FEBS J. 2019. PMID: 30768849 Free PMC article.
-
Mutations in repeating structural motifs of tropomyosin cause gain of function in skeletal muscle myopathy patients.Hum Mol Genet. 2013 Dec 15;22(24):4978-87. doi: 10.1093/hmg/ddt345. Epub 2013 Jul 25. Hum Mol Genet. 2013. PMID: 23886664 Free PMC article.
-
Congenital myopathies: diseases of the actin cytoskeleton.J Pathol. 2004 Nov;204(4):407-17. doi: 10.1002/path.1648. J Pathol. 2004. PMID: 15495263 Review.
Cited by
-
NEB mutations disrupt the super-relaxed state of myosin and remodel the muscle metabolic proteome in nemaline myopathy.Acta Neuropathol Commun. 2022 Dec 17;10(1):185. doi: 10.1186/s40478-022-01491-9. Acta Neuropathol Commun. 2022. PMID: 36528760 Free PMC article.
-
A two-segment model for thin filament architecture in skeletal muscle.Nat Rev Mol Cell Biol. 2013 Feb;14(2):113-9. doi: 10.1038/nrm3510. Epub 2013 Jan 9. Nat Rev Mol Cell Biol. 2013. PMID: 23299957 Free PMC article.
-
Troponin and a Myopathy-Linked Mutation in TPM3 Inhibit Cofilin-2-Induced Thin Filament Depolymerization.Int J Mol Sci. 2023 Nov 17;24(22):16457. doi: 10.3390/ijms242216457. Int J Mol Sci. 2023. PMID: 38003645 Free PMC article.
-
Poorly understood aspects of striated muscle contraction.Biomed Res Int. 2015;2015:245154. doi: 10.1155/2015/245154. Epub 2015 Apr 16. Biomed Res Int. 2015. PMID: 25961006 Free PMC article. Review.
-
Thin filament dysfunctions caused by mutations in tropomyosin Tpm3.12 and Tpm1.1.J Muscle Res Cell Motil. 2020 Mar;41(1):39-53. doi: 10.1007/s10974-019-09532-y. Epub 2019 Jul 3. J Muscle Res Cell Motil. 2020. PMID: 31270709 Free PMC article. Review.
References
-
- Lehman W., Craig R. Tropomyosin and the steric mechanism of muscle regulation. Adv. Exp. Med. Biol. 2008;644:95–109. doi:10.1007/978-0-387-85766-4_8. - DOI - PubMed
-
- Ochala J. Thin filament proteins mutations associated with skeletal myopathies: defective regulation of muscle contraction. J. Mol. Med. 2008;86:1197–1204. doi:10.1007/s00109-008-0380-9. - DOI - PubMed
-
- Ochala J., Li M., Ohlsson M., Oldfors A., Larsson L. Defective regulation of contractile function in muscle fibres carrying an E41K beta-tropomyosin mutation. J. Physiol. 2008;586:2993–3004. doi:10.1113/jphysiol.2008.153650. - DOI - PMC - PubMed
-
- Ottenheijm C.A., Lawlor M.W., Stienen G.J., Granzier H., Beggs A.H. Changes in cross-bridge cycling underlie muscle weakness in patients with tropomyosin 3-based myopathy. Hum. Mol. Genet. 2011;20:2015–2025. doi:10.1093/hmg/ddr084. - DOI - PMC - PubMed
-
- Sanoudou D., Beggs A.H. Clinical and genetic heterogeneity in nemaline myopathy—a disease of skeletal muscle thin filaments. Trends Mol. Med. 2001;7:362–368. doi:10.1016/S1471-4914(01)02089-5. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
