

Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis
- PMID: 22801503
- PMCID: PMC3575525
- DOI: 10.1038/nature11280
Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis
Abstract
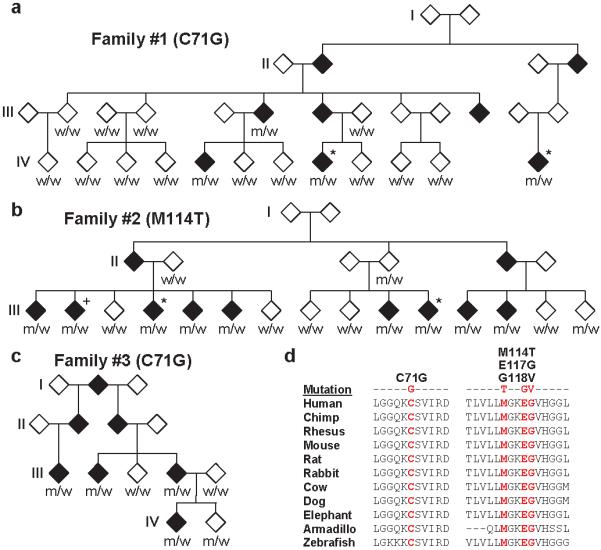
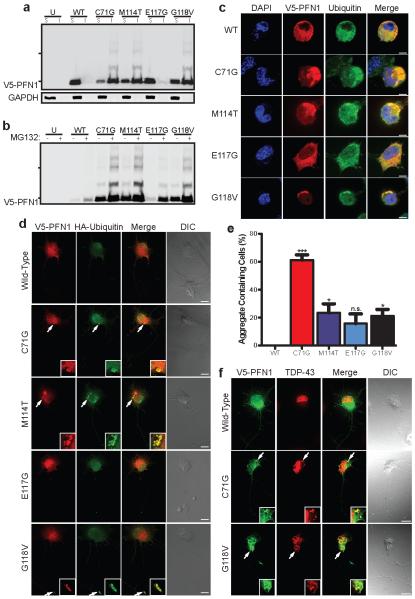
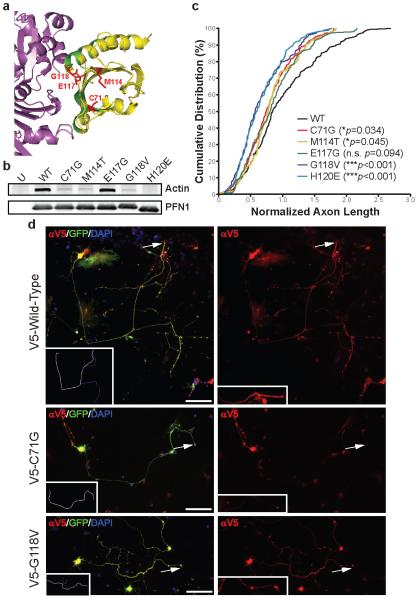
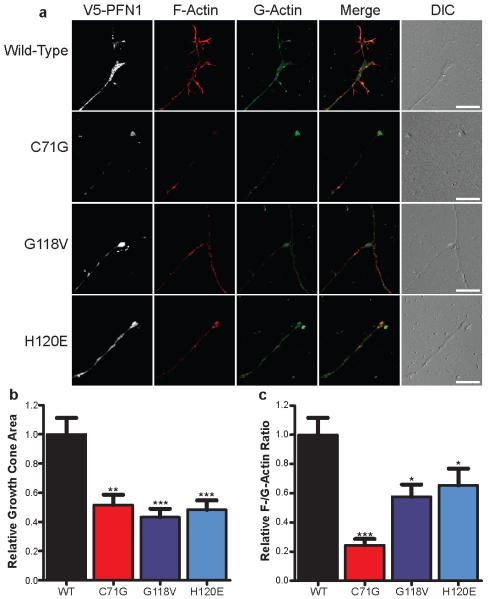
Amyotrophic lateral sclerosis (ALS) is a late-onset neurodegenerative disorder resulting from motor neuron death. Approximately 10% of cases are familial (FALS), typically with a dominant inheritance mode. Despite numerous advances in recent years, nearly 50% of FALS cases have unknown genetic aetiology. Here we show that mutations within the profilin 1 (PFN1) gene can cause FALS. PFN1 is crucial for the conversion of monomeric (G)-actin to filamentous (F)-actin. Exome sequencing of two large ALS families showed different mutations within the PFN1 gene. Further sequence analysis identified 4 mutations in 7 out of 274 FALS cases. Cells expressing PFN1 mutants contain ubiquitinated, insoluble aggregates that in many cases contain the ALS-associated protein TDP-43. PFN1 mutants also display decreased bound actin levels and can inhibit axon outgrowth. Furthermore, primary motor neurons expressing mutant PFN1 display smaller growth cones with a reduced F/G-actin ratio. These observations further document that cytoskeletal pathway alterations contribute to ALS pathogenesis.
Figures
References
-
- Kabashi E, et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40:572–574. - PubMed
-
- Kwiatkowski TJ, Jr., et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–1208. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
