

Explorations of catalytic domains in non-ribosomal peptide synthetase enzymology
- PMID: 22802156
- PMCID: PMC4807874
- DOI: 10.1039/c2np20025b
Explorations of catalytic domains in non-ribosomal peptide synthetase enzymology
Abstract
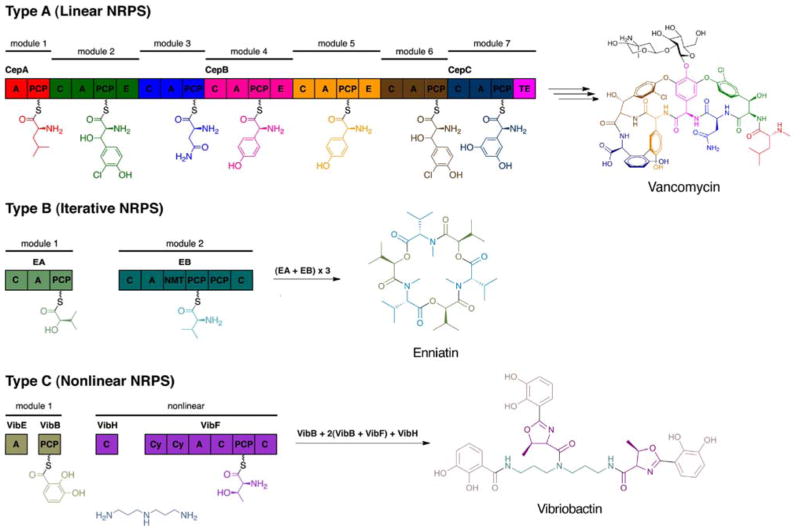
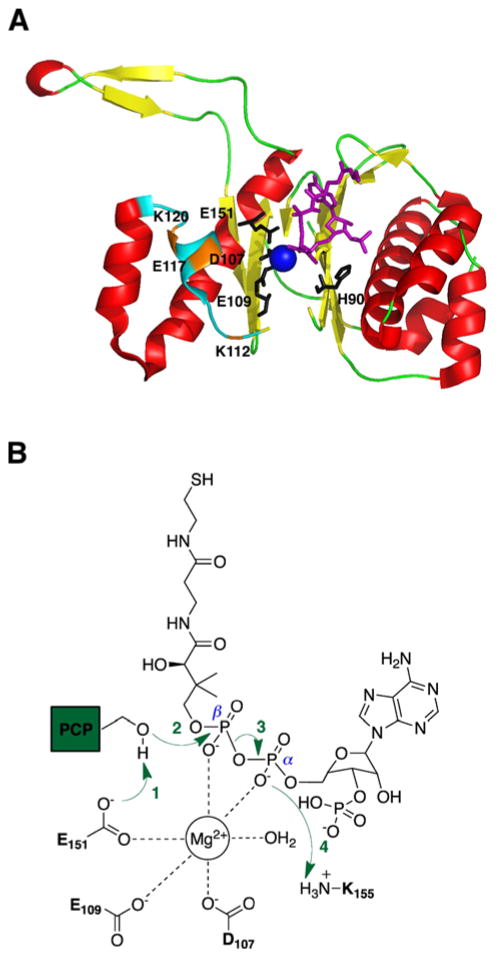
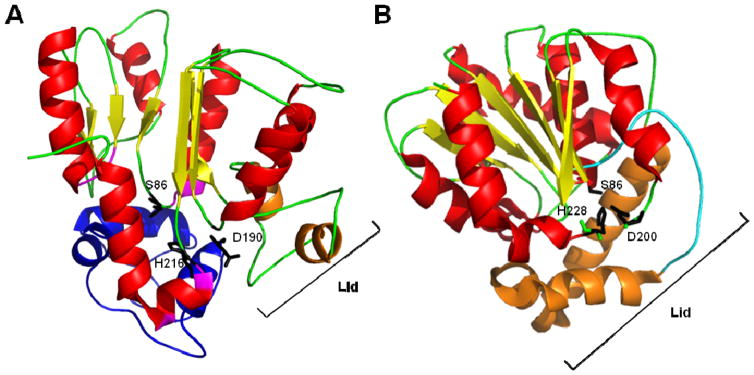
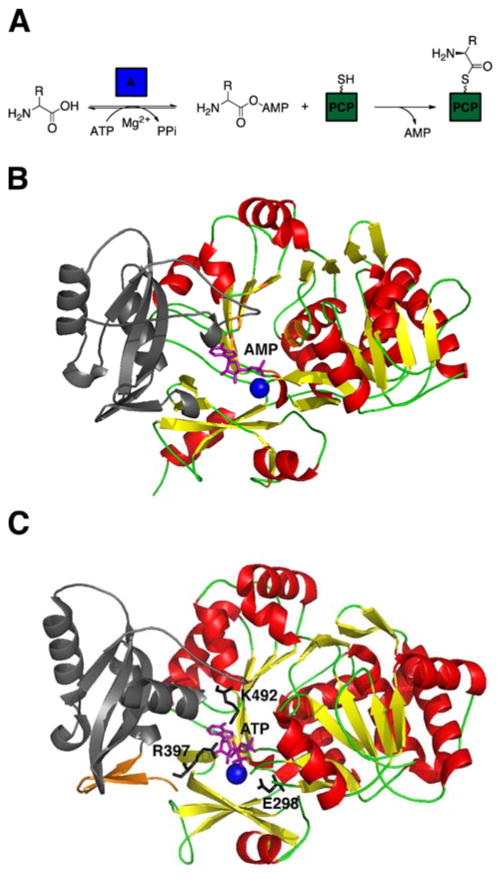
Many pharmaceuticals on the market today belong to a large class of natural products called nonribosomal peptides (NRPs). Originating from bacteria and fungi, these peptide-based natural products consist not only of the 20 canonical L-amino acids, but also non-proteinogenic amino acids, heterocyclic rings, sugars, and fatty acids, generating tremendous chemical diversity. As a result, these secondary metabolites exhibit a broad array of bioactivity, ranging from antimicrobial to anticancer. The biosynthesis of these complex compounds is carried out by large multimodular megaenzymes called nonribosomal peptide synthetases (NRPSs). Each module is responsible for incorporation of a monomeric unit into the natural product peptide and is composed of individual domains that perform different catalytic reactions. Biochemical and bioinformatic investigations of these enzymes have uncovered the key principles of NRP synthesis, expanding the pharmaceutical potential of their enzymatic processes. Progress has been made in the manipulation of this biosynthetic machinery to develop new chemoenzymatic approaches for synthesizing novel pharmaceutical agents with increased potency. This review focuses on the recent discoveries and breakthroughs in the structural elucidation, molecular mechanism, and chemical biology underlying the discrete domains within NRPSs.
Figures

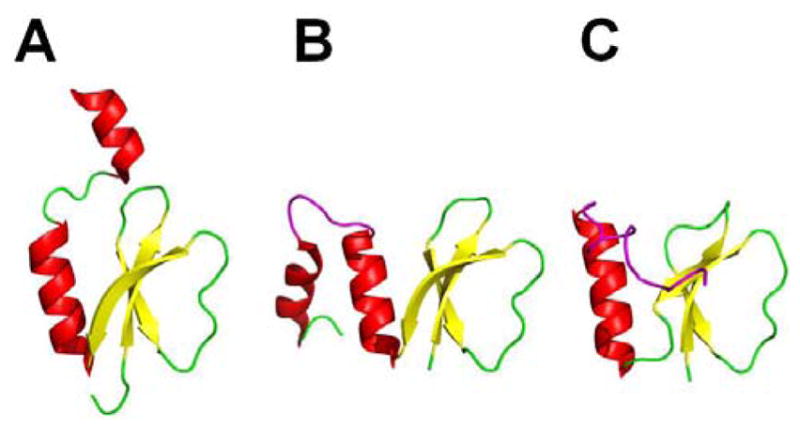
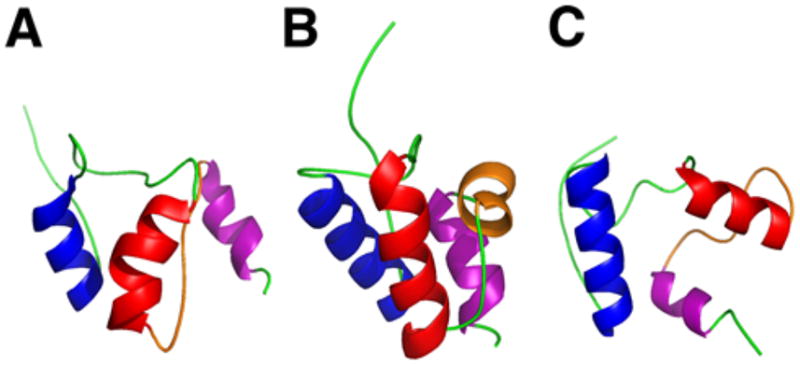
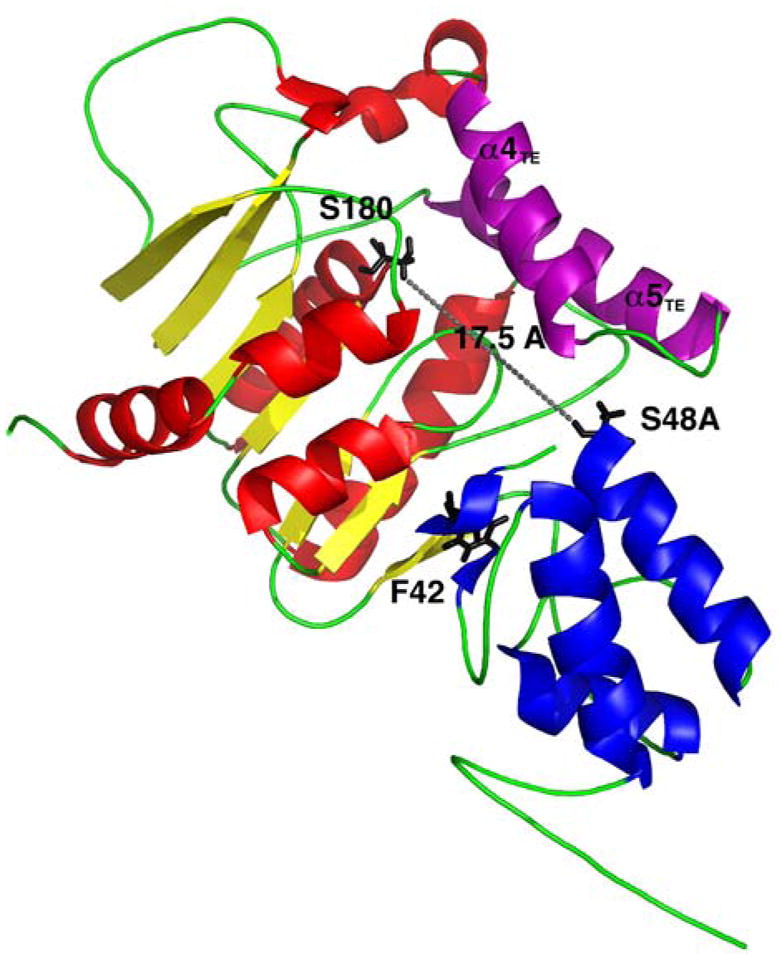
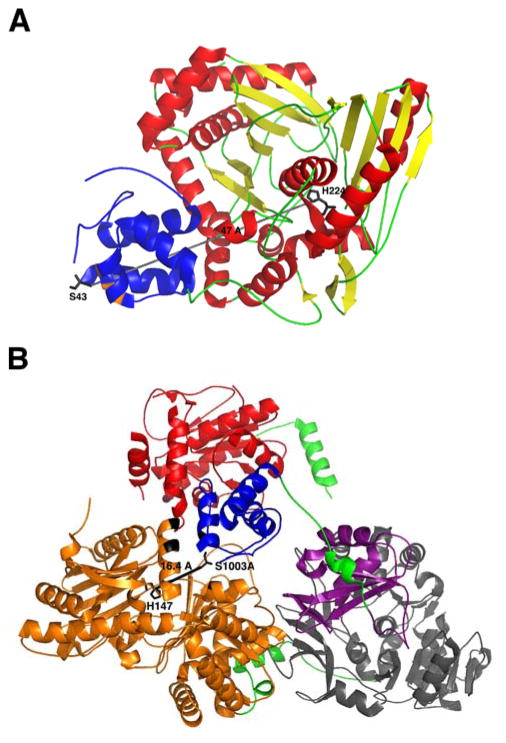

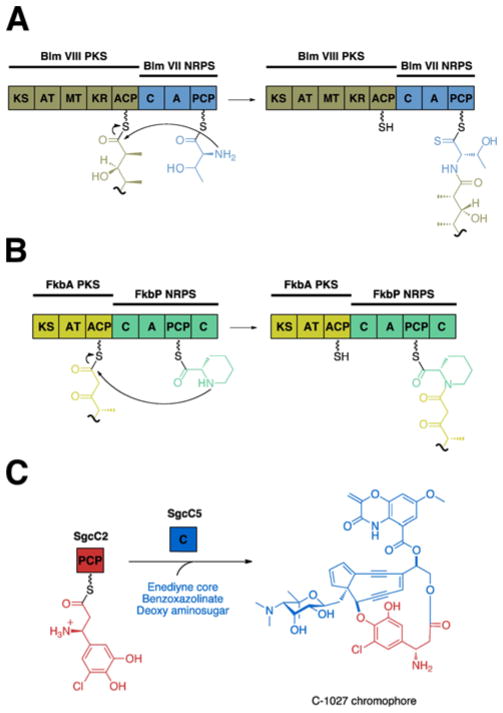
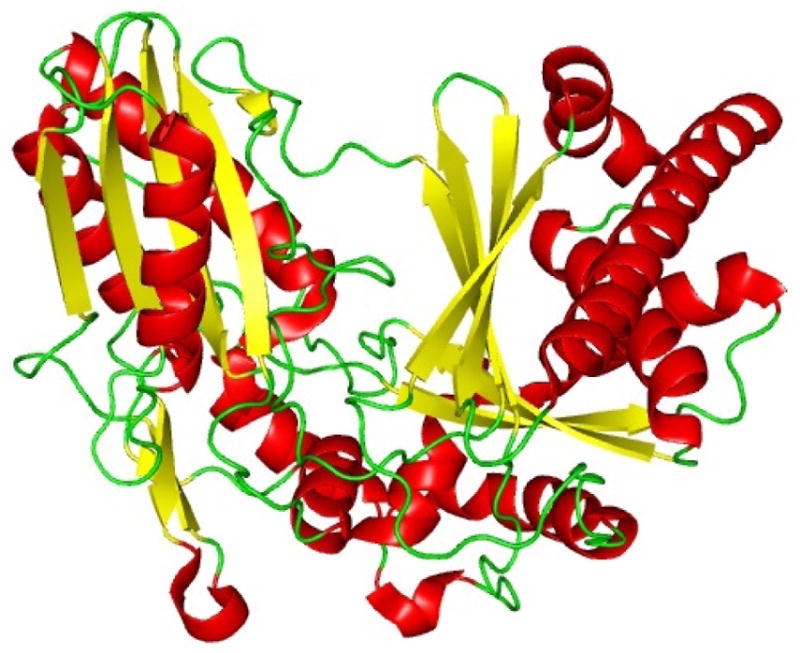
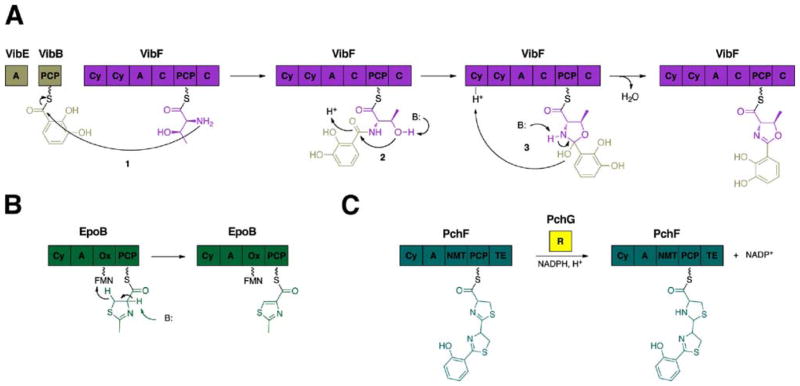

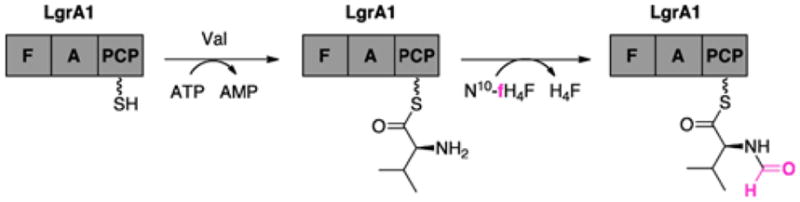
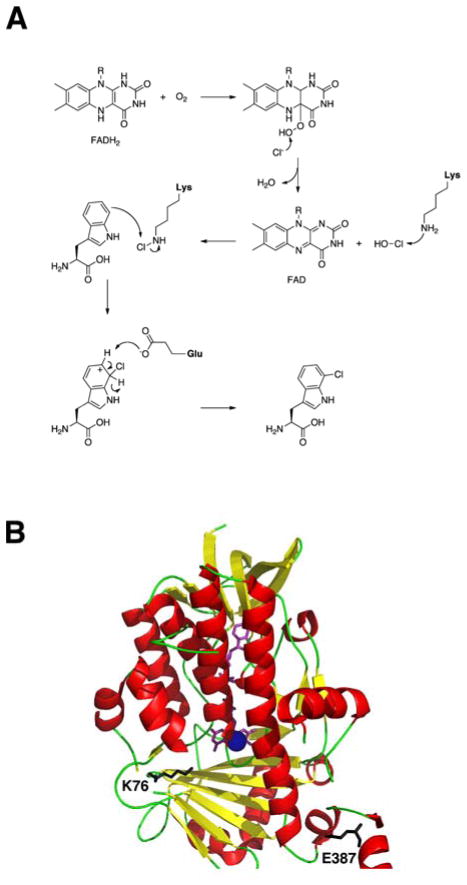
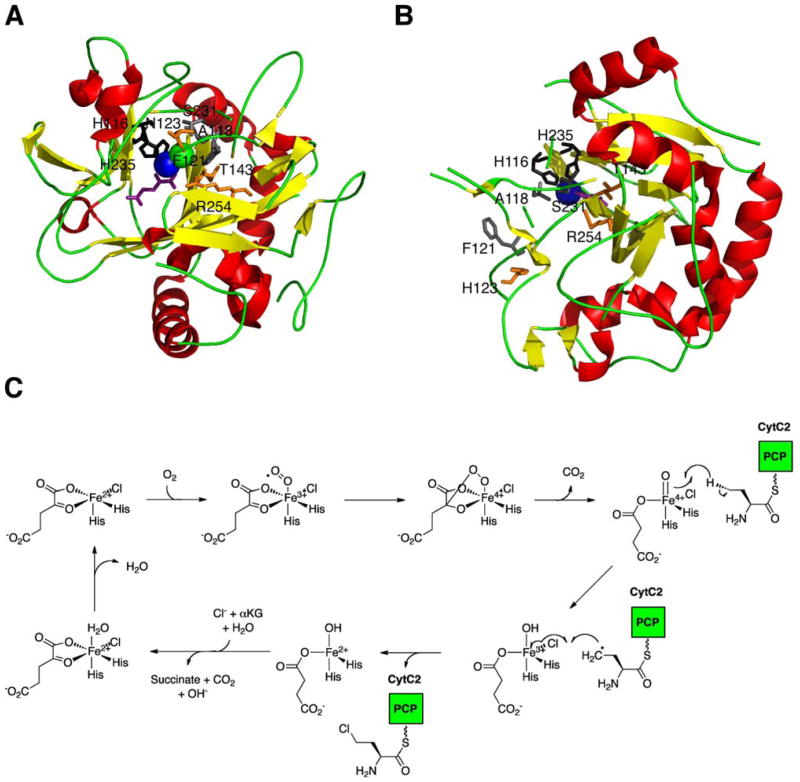
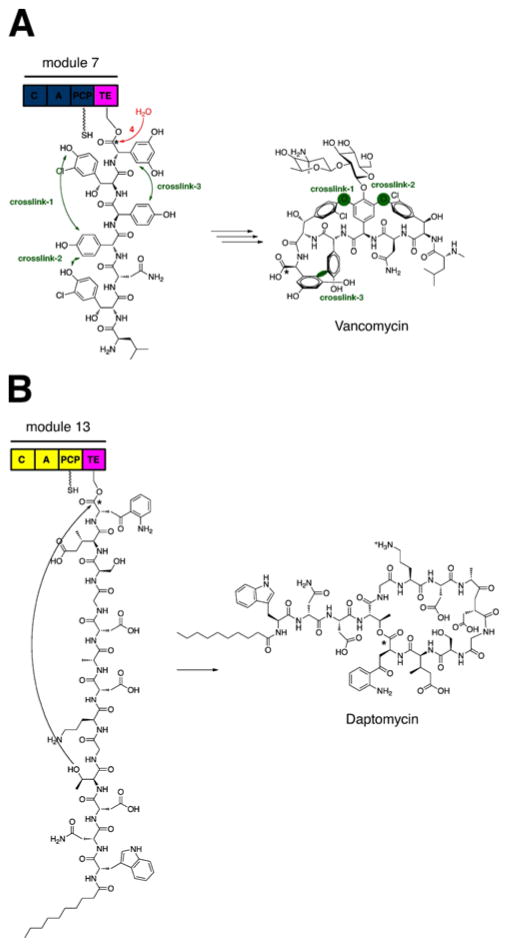
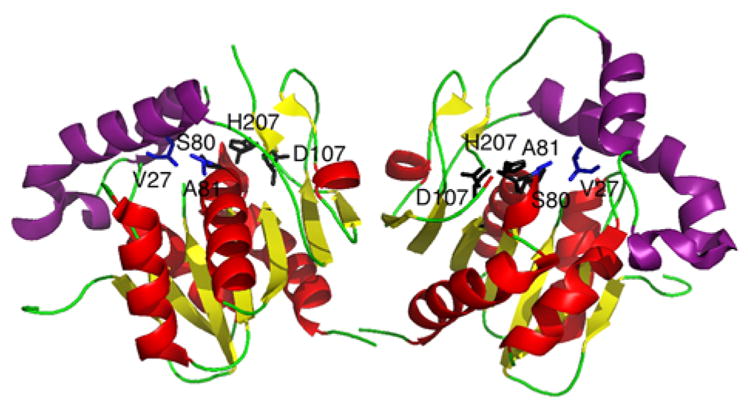
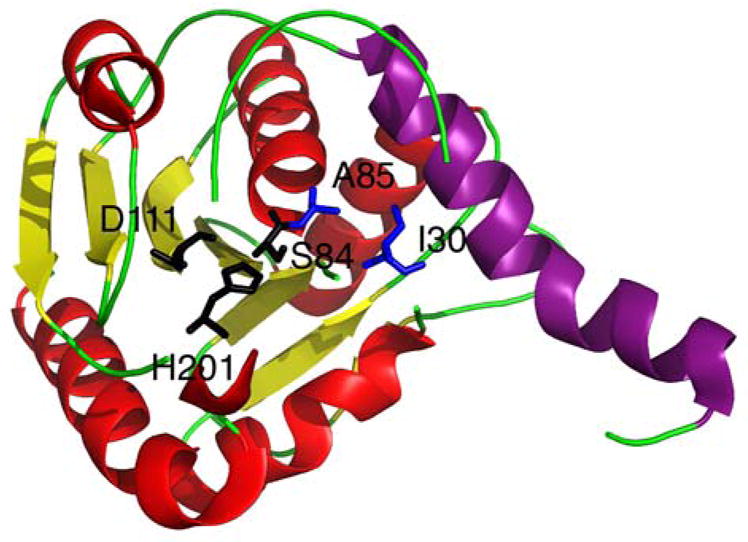
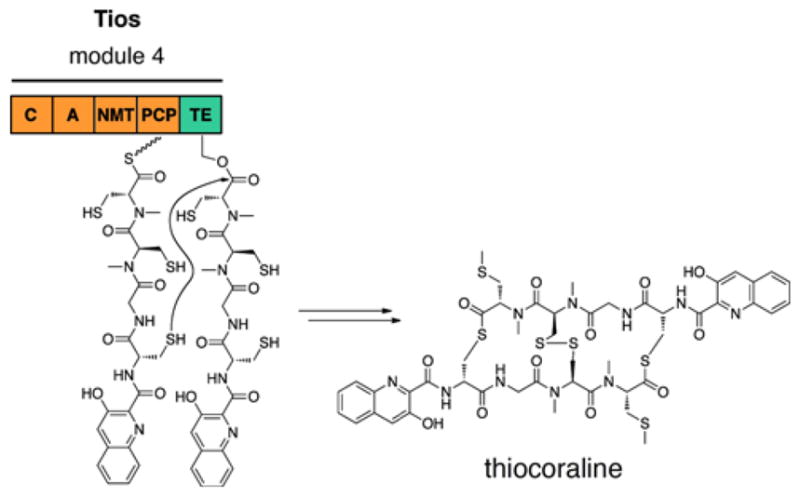

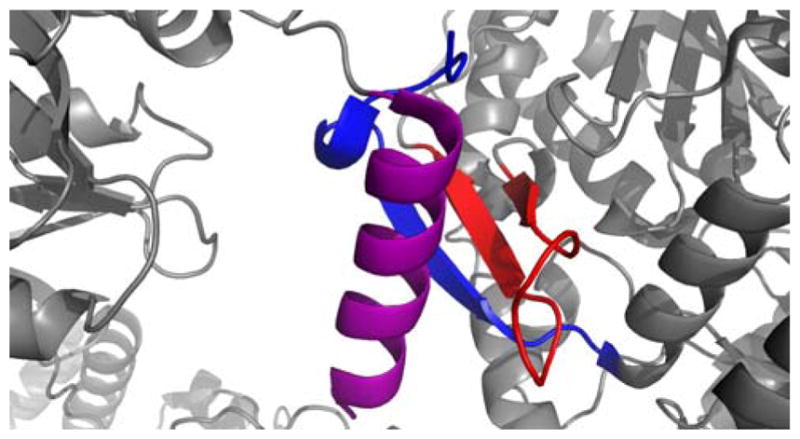
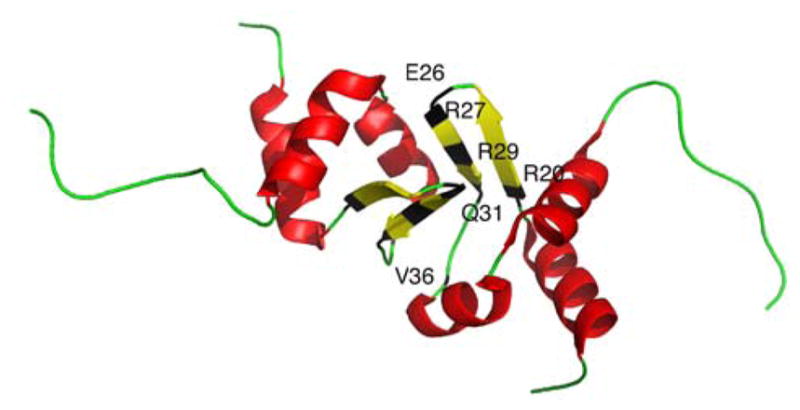
References
-
- Finking R, Marahiel MA. Annu Rev Microbiol. 2004;58:453–488. - PubMed
-
- Cane DE, Walsh CT, Khosla C. Science. 1998;282:63–68. - PubMed
-
- Stein T, Vater J, Kruft V, Otto A, Wittmann-Liebold B, Franke P, Panico M, McDowell R, Morris HR. J Biol Chem. 1996;271:15428–15435. - PubMed
-
- Weber G, Schorgendorfer K, Schneider-Scherzer E, Leitner E. Curr Genet. 1994;26:120–125. - PubMed
-
- Jirakkakul J, Punya J, Pongpattanakitshote S, Paungmoung P, Vorapreeda N, Tachaleat A, Klomnara C, Tanticharoen M, Cheevadhanarak S. Microbiology. 2008;154:995–1006. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous