

Role of the lysophospholipid mediators lysophosphatidic acid and sphingosine 1-phosphate in lung fibrosis
- PMID: 22802282
- PMCID: PMC5455616
- DOI: 10.1513/pats.201201-005AW
Role of the lysophospholipid mediators lysophosphatidic acid and sphingosine 1-phosphate in lung fibrosis
Abstract
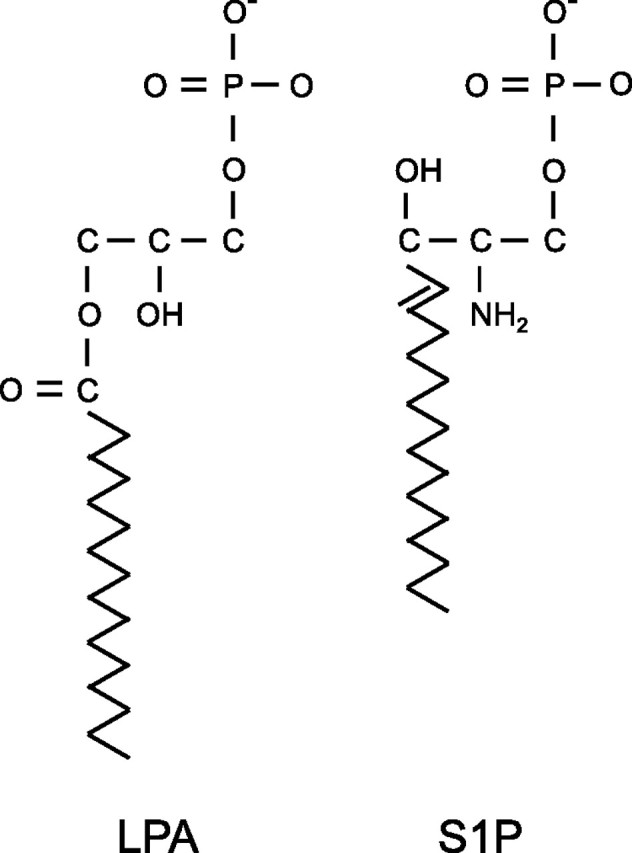
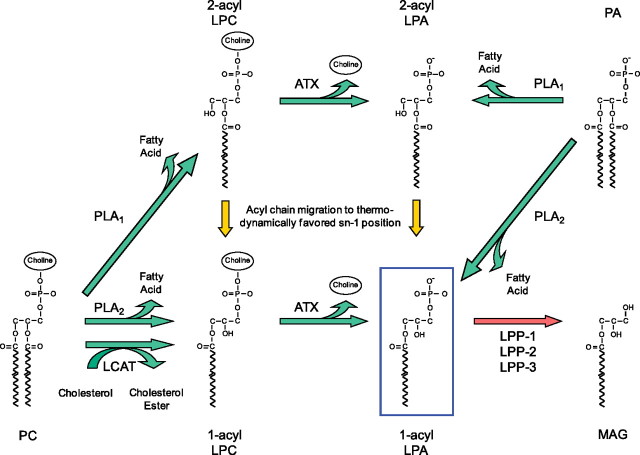
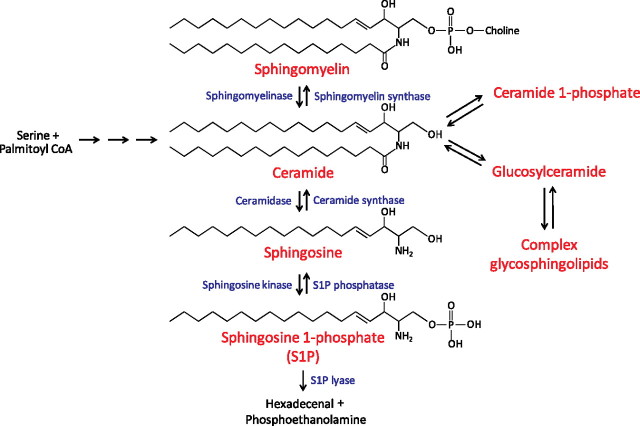
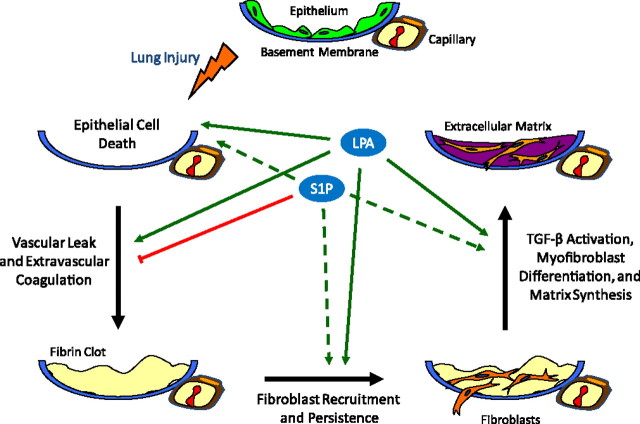
Aberrant wound healing responses to lung injury are believed to contribute to fibrotic lung diseases, such as idiopathic pulmonary fibrosis (IPF). The lysophospholipids lysophosphatidic acid (LPA) and sphingosine 1-phosphate (S1P), by virtue of their ability to mediate many basic cellular functions, including survival, proliferation, migration, and contraction, can influence many of the biological processes involved in wound healing. Accordingly, recent investigations indicate that LPA and S1P may play critical roles in regulating the development of lung fibrosis. Here we review the evidence indicating that LPA and S1P regulate pulmonary fibrosis and the potential mechanisms through which these lysophospholipids may influence fibrogenesis induced by lung injury.
Figures
References
-
- Ley B, Collard HR, King TE. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2010;183:431–440. - PubMed
-
- Selman M, Pardo A. Role of epithelial cells in idiopathic pulmonary fibrosis: from innocent targets to serial killers. Proc Am Thorac Soc 2006;3:364–372. - PubMed
-
- Gharaee-Kermani M, Hu B, Phan SH, Gyetko MR. Recent advances in molecular targets and treatment of idiopathic pulmonary fibrosis: focus on TGFbeta signaling and the myofibroblast. Curr Med Chem 2009;16:1400–1417. - PubMed
-
- Antoniou KM, Pataka A, Bouros D, Siafakas NM. Pathogenetic pathways and novel pharmacotherapeutic targets in idiopathic pulmonary fibrosis. Pulm Pharmacol Ther 2007;20:453–461. - PubMed
-
- Coward WR, Saini G, Jenkins G. The pathogenesis of idiopathic pulmonary fibrosis. Ther Adv Respir Dis 2010;4:367–388. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous