

Alzheimer's disease Aβ assemblies mediating rapid disruption of synaptic plasticity and memory
- PMID: 22805374
- PMCID: PMC3502131
- DOI: 10.1186/1756-6606-5-25
Alzheimer's disease Aβ assemblies mediating rapid disruption of synaptic plasticity and memory
Abstract
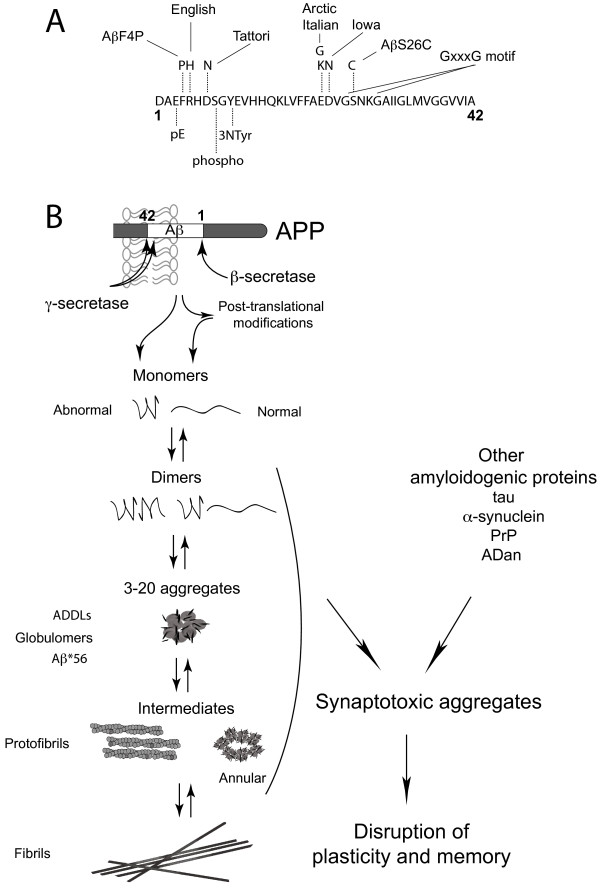
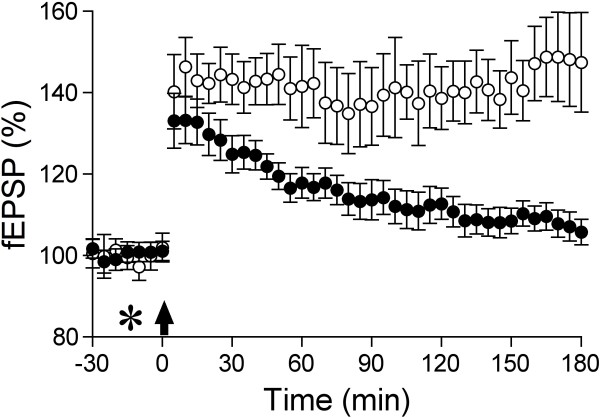
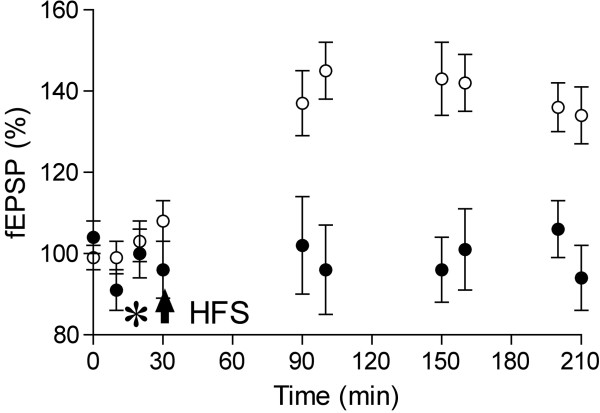
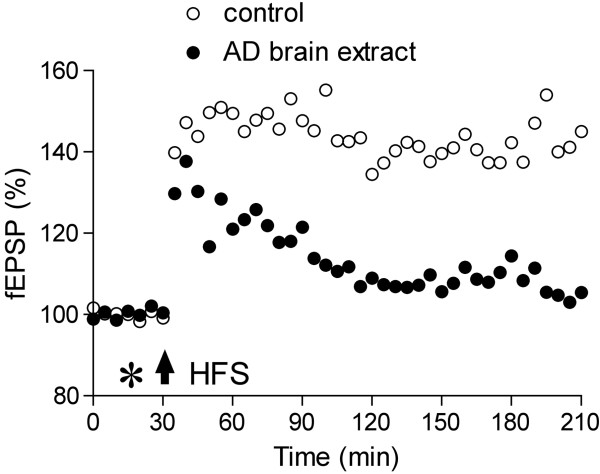
Alzheimer's disease (AD) is characterized by episodic memory impairment that often precedes clinical diagnosis by many years. Probing the mechanisms of such impairment may provide much needed means of diagnosis and therapeutic intervention at an early, pre-dementia, stage. Prior to the onset of significant neurodegeneration, the structural and functional integrity of synapses in mnemonic circuitry is severely compromised in the presence of amyloidosis. This review examines recent evidence evaluating the role of amyloid-ß protein (Aβ) in causing rapid disruption of synaptic plasticity and memory impairment. We evaluate the relative importance of different sizes and conformations of Aβ, including monomer, oligomer, protofibril and fibril. We pay particular attention to recent controversies over the relevance to the pathophysiology of AD of different water soluble Aβ aggregates and the importance of cellular prion protein in mediating their effects. Current data are consistent with the view that both low-n oligomers and larger soluble assemblies present in AD brain, some of them via a direct interaction with cellular prion protein, cause synaptic memory failure. At the two extremes of aggregation, monomers and fibrils appear to act in vivo both as sources and sinks of certain metastable conformations of soluble aggregates that powerfully disrupt synaptic plasticity. The same principle appears to apply to other synaptotoxic amyloidogenic proteins including tau, α-synuclein and prion protein.
Figures
References
-
- Roberts GW, Lofthouse R, Allsop D, Landon M, Kidd M, Prusiner SB, Crow TJ. CNS amyloid proteins in neurodegenerative diseases. Neurology. 1988;38:1534–1540. - PubMed
-
- Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. - PubMed
-
- Palop JJ, Chin J, Mucke L. A network dysfunction perspective on neurodegenerative diseases. Nature. 2006;443:768–773. - PubMed
-
- Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. - PubMed
-
- Schneider JA, Arvanitakis Z, Bang W, Bennett DA. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology. 2007;69:2197–2204. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
