

MGMT methylation analysis of glioblastoma on the Infinium methylation BeadChip identifies two distinct CpG regions associated with gene silencing and outcome, yielding a prediction model for comparisons across datasets, tumor grades, and CIMP-status
- PMID: 22810491
- PMCID: PMC3444709
- DOI: 10.1007/s00401-012-1016-2
MGMT methylation analysis of glioblastoma on the Infinium methylation BeadChip identifies two distinct CpG regions associated with gene silencing and outcome, yielding a prediction model for comparisons across datasets, tumor grades, and CIMP-status
Erratum in
- Acta Neuropathol. 2013 Jul;126(1):159
Abstract
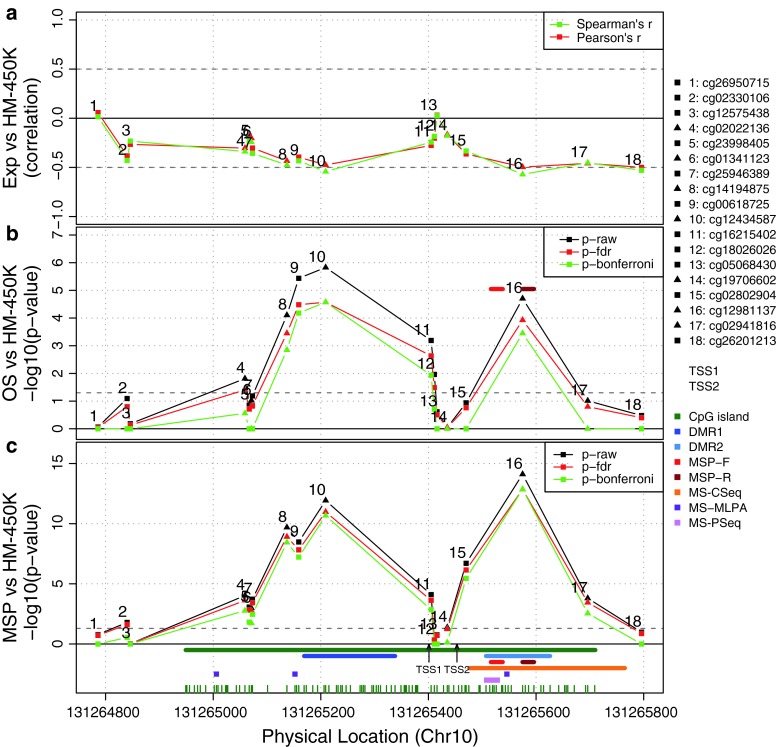
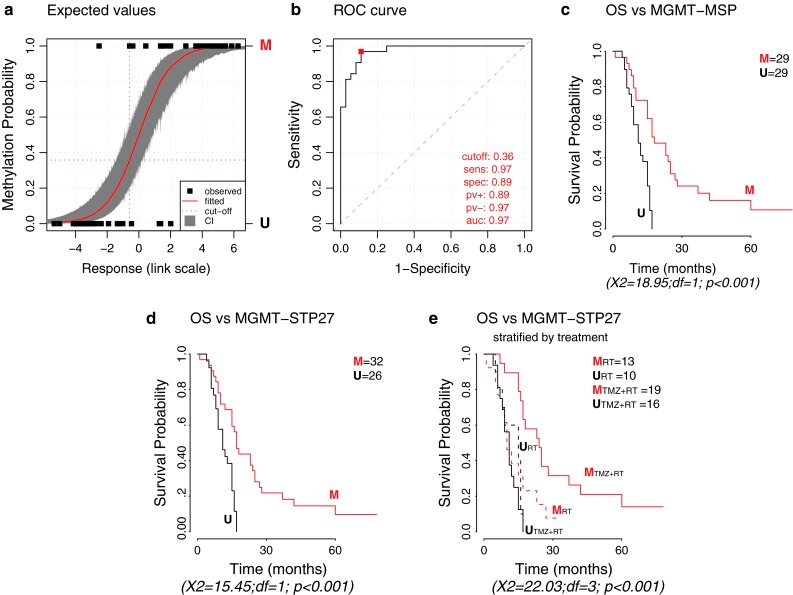
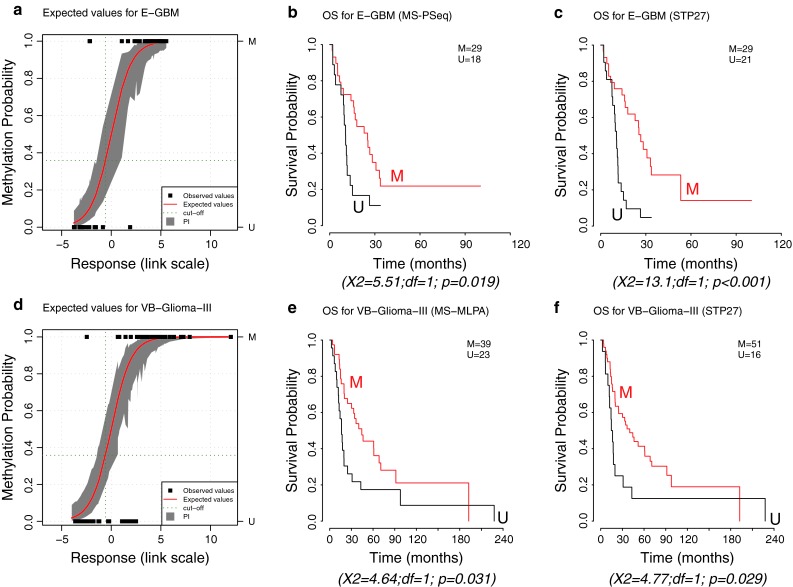
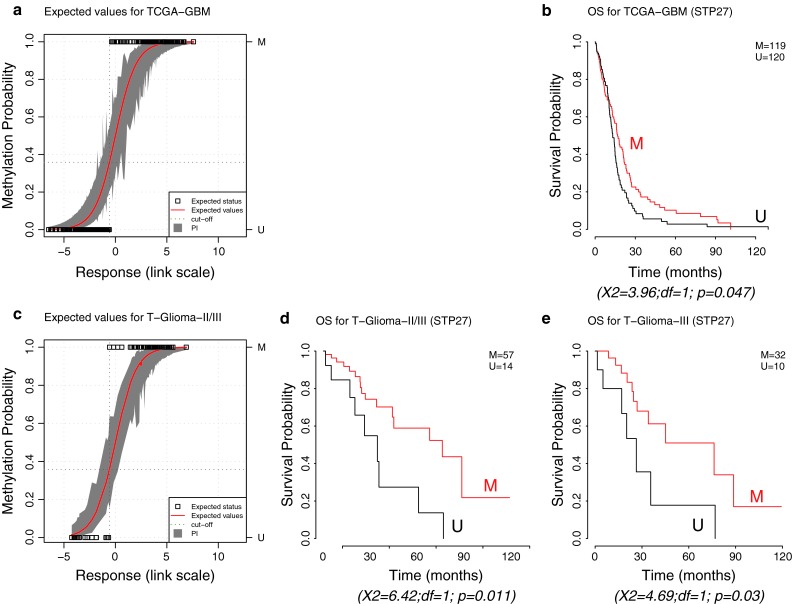
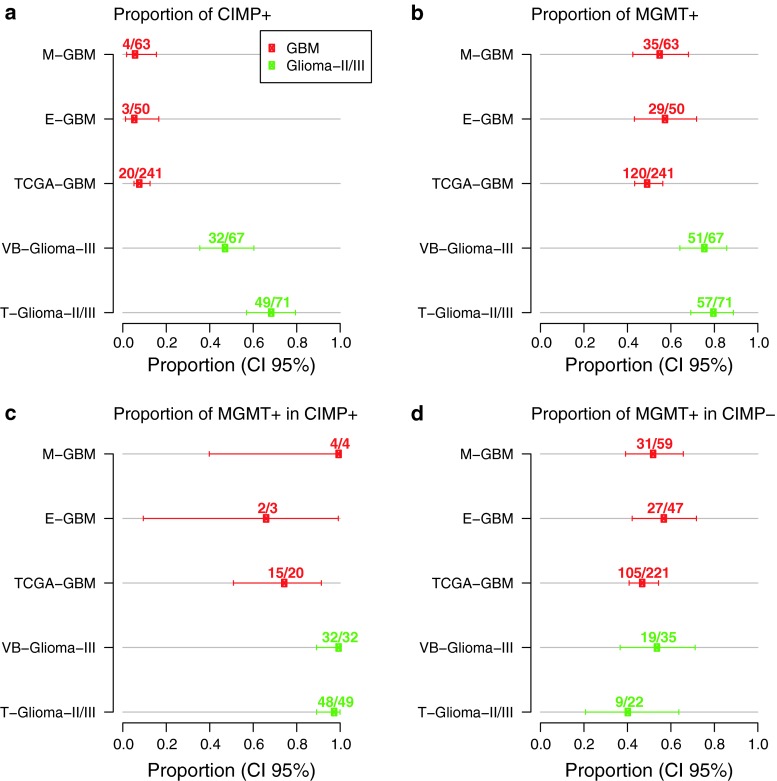
The methylation status of the O(6)-methylguanine-DNA methyltransferase (MGMT) gene is an important predictive biomarker for benefit from alkylating agent therapy in glioblastoma. Recent studies in anaplastic glioma suggest a prognostic value for MGMT methylation. Investigation of pathogenetic and epigenetic features of this intriguingly distinct behavior requires accurate MGMT classification to assess high throughput molecular databases. Promoter methylation-mediated gene silencing is strongly dependent on the location of the methylated CpGs, complicating classification. Using the HumanMethylation450 (HM-450K) BeadChip interrogating 176 CpGs annotated for the MGMT gene, with 14 located in the promoter, two distinct regions in the CpG island of the promoter were identified with high importance for gene silencing and outcome prediction. A logistic regression model (MGMT-STP27) comprising probes cg12434587 [corrected] and cg12981137 provided good classification properties and prognostic value (kappa = 0.85; log-rank p < 0.001) using a training-set of 63 glioblastomas from homogenously treated patients, for whom MGMT methylation was previously shown to be predictive for outcome based on classification by methylation-specific PCR. MGMT-STP27 was successfully validated in an independent cohort of chemo-radiotherapy-treated glioblastoma patients (n = 50; kappa = 0.88; outcome, log-rank p < 0.001). Lower prevalence of MGMT methylation among CpG island methylator phenotype (CIMP) positive tumors was found in glioblastomas from The Cancer Genome Atlas than in low grade and anaplastic glioma cohorts, while in CIMP-negative gliomas MGMT was classified as methylated in approximately 50 % regardless of tumor grade. The proposed MGMT-STP27 prediction model allows mining of datasets derived on the HM-450K or HM-27K BeadChip to explore effects of distinct epigenetic context of MGMT methylation suspected to modulate treatment resistance in different tumor types.
Figures

References
-
- Burnham KP, Anderson DR. Model selection and multimodel inference: a practical information theoretic approach. New York: Springer-Verlag; 2002.
-
- Collett D. Modelling binary data. London: Chapman & Hall/CRC; 2002.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
