

Control of sleep and wakefulness
- PMID: 22811426
- PMCID: PMC3621793
- DOI: 10.1152/physrev.00032.2011
Control of sleep and wakefulness
Abstract
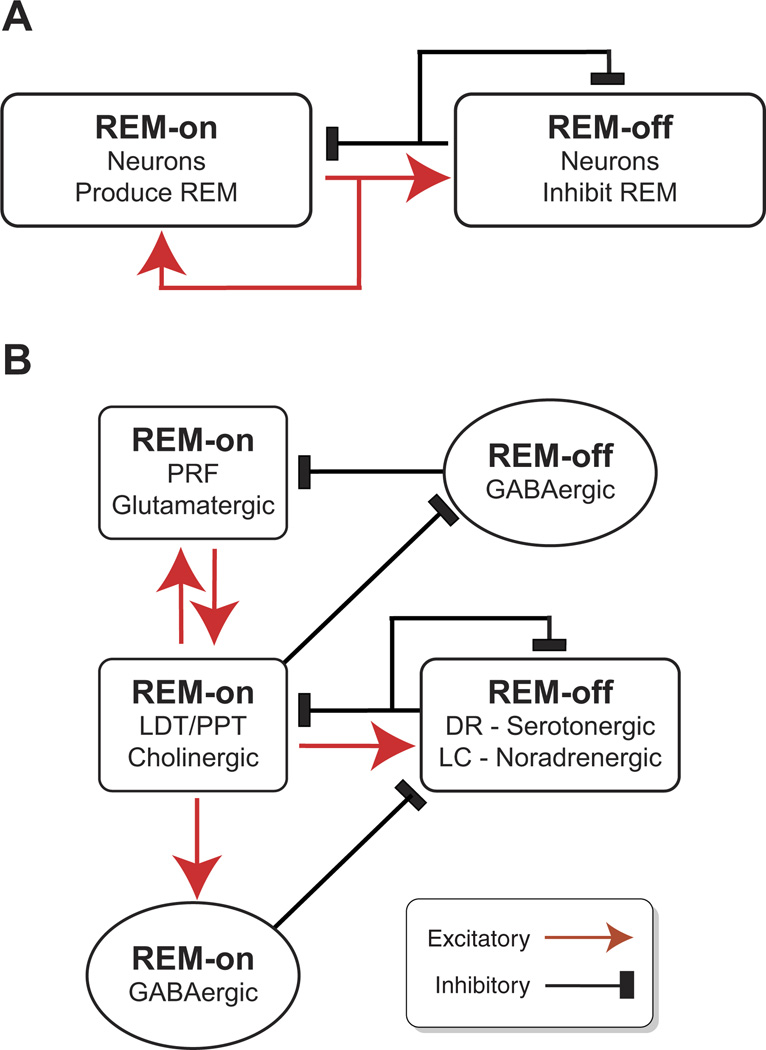
This review summarizes the brain mechanisms controlling sleep and wakefulness. Wakefulness promoting systems cause low-voltage, fast activity in the electroencephalogram (EEG). Multiple interacting neurotransmitter systems in the brain stem, hypothalamus, and basal forebrain converge onto common effector systems in the thalamus and cortex. Sleep results from the inhibition of wake-promoting systems by homeostatic sleep factors such as adenosine and nitric oxide and GABAergic neurons in the preoptic area of the hypothalamus, resulting in large-amplitude, slow EEG oscillations. Local, activity-dependent factors modulate the amplitude and frequency of cortical slow oscillations. Non-rapid-eye-movement (NREM) sleep results in conservation of brain energy and facilitates memory consolidation through the modulation of synaptic weights. Rapid-eye-movement (REM) sleep results from the interaction of brain stem cholinergic, aminergic, and GABAergic neurons which control the activity of glutamatergic reticular formation neurons leading to REM sleep phenomena such as muscle atonia, REMs, dreaming, and cortical activation. Strong activation of limbic regions during REM sleep suggests a role in regulation of emotion. Genetic studies suggest that brain mechanisms controlling waking and NREM sleep are strongly conserved throughout evolution, underscoring their enormous importance for brain function. Sleep disruption interferes with the normal restorative functions of NREM and REM sleep, resulting in disruptions of breathing and cardiovascular function, changes in emotional reactivity, and cognitive impairments in attention, memory, and decision making.
Conflict of interest statement
No conflicts of interest, financial or otherwise, are declared by the authors.
Figures












References
-
- Abel GG, Murphy WD, Becker JV, Bitar A. Women’s vaginal responses during REM sleep. J Sex Marital Ther. 1979;5:5–14. - PubMed
-
- Achermann P, Borbely AA. Mathematical models of sleep regulation. Front Biosci. 2003;8 s683-1945-s693. - PubMed
-
- Adamantidis A, Salvert D, Goutagny R, Lakaye B, Gervasoni D, Grisar T, Luppi PH, Fort P. Sleep architecture of the melanin-concentrating hormone receptor 1-knockout mice. Eur J Neurosci. 2008;27:1793–1800. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical