

Fast and accurate read alignment for resequencing
- PMID: 22811546
- PMCID: PMC3436849
- DOI: 10.1093/bioinformatics/bts450
Fast and accurate read alignment for resequencing
Abstract
Motivation: Next-generation sequence analysis has become an important task both in laboratory and clinical settings. A key stage in the majority sequence analysis workflows, such as resequencing, is the alignment of genomic reads to a reference genome. The accurate alignment of reads with large indels is a computationally challenging task for researchers.
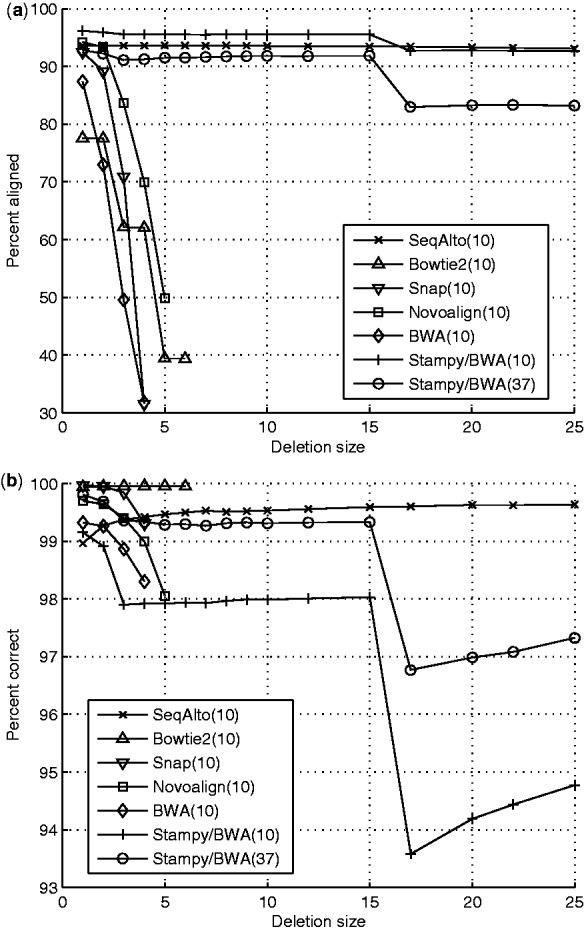
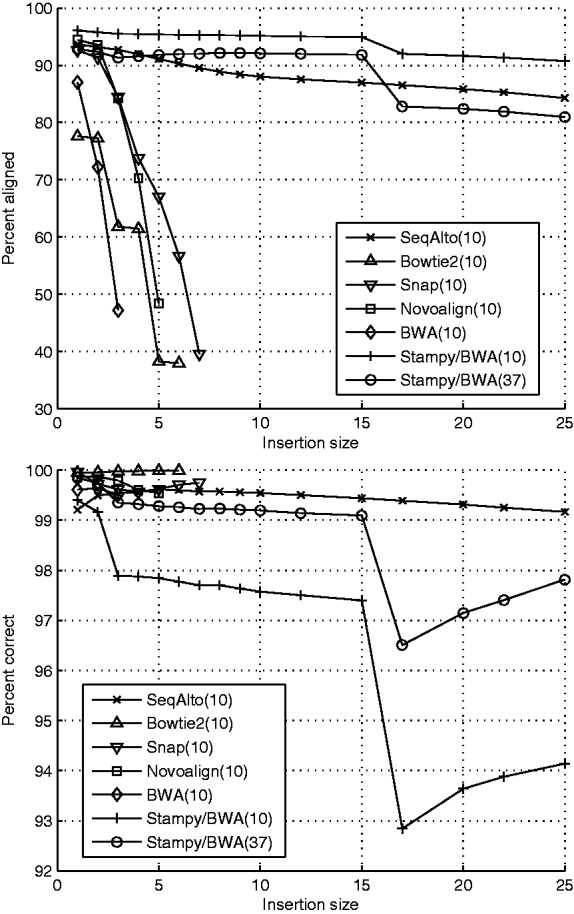
Results: We introduce SeqAlto as a new algorithm for read alignment. For reads longer than or equal to 100 bp, SeqAlto is up to 10 × faster than existing algorithms, while retaining high accuracy and the ability to align reads with large (up to 50 bp) indels. This improvement in efficiency is particularly important in the analysis of future sequencing data where the number of reads approaches many billions. Furthermore, SeqAlto uses less than 8 GB of memory to align against the human genome. SeqAlto is benchmarked against several existing tools with both real and simulated data.
Availability: Linux and Mac OS X binaries free for academic use are available at http://www.stanford.edu/group/wonglab/seqalto
Contact: whwong@stanford.edu.
Figures
References
-
- Baeza-yates RA, Perleberg CH. Combinatorial Pattern Matching, Third Annual Symposium. Springer-Verlag; 1992. Fast and practical approximate string matching; pp. 185–192.
-
- Burrows M, Wheeler DJ. A block-sorting lossless data compression algorithm. HP Labs Technical Reports, SRC-RR-124. 1994
-
- David M, et al. SHRiMP2: sensitive yet practical short read mapping. Bioinformatics. 2011;27:1011–1012. - PubMed
-
- Ferragina P, Manzini G. In Proc. 41st IEEE Symposium on Foundations of Computer Science (FOCS). Redondo, Beach, CA, USA. Redondo Beach, CA, USA: 2000. Opportunistic data structures with applications.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
