

A mutation in TNNC1-encoded cardiac troponin C, TNNC1-A31S, predisposes to hypertrophic cardiomyopathy and ventricular fibrillation
- PMID: 22815480
- PMCID: PMC3442518
- DOI: 10.1074/jbc.M112.377713
A mutation in TNNC1-encoded cardiac troponin C, TNNC1-A31S, predisposes to hypertrophic cardiomyopathy and ventricular fibrillation
Abstract
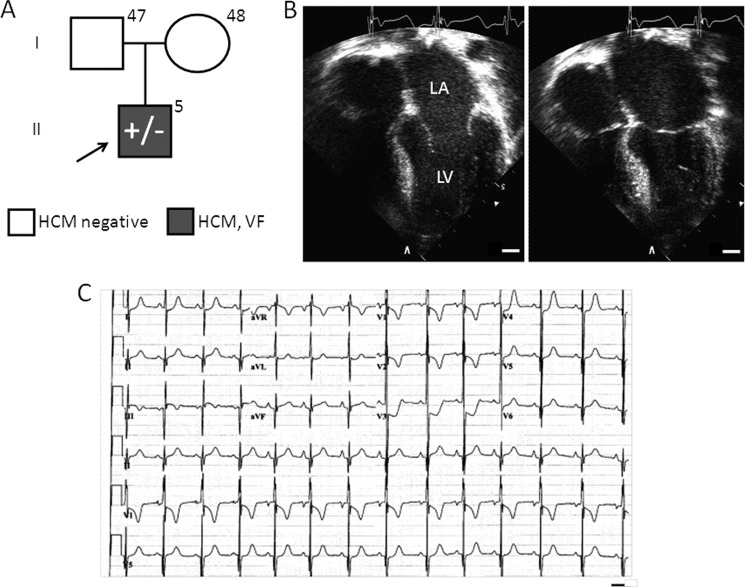
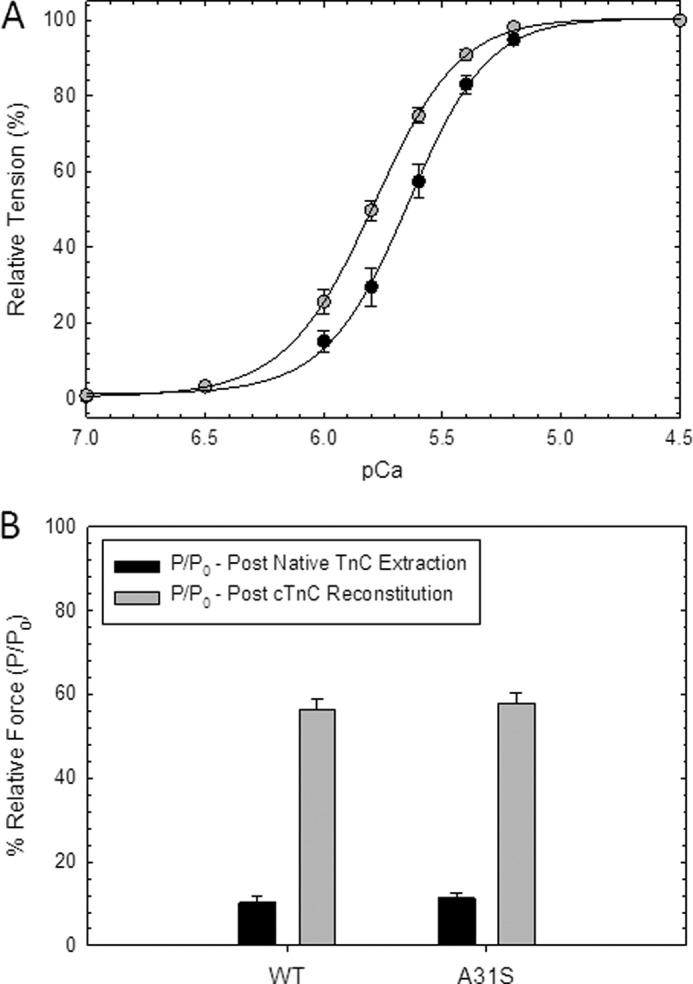
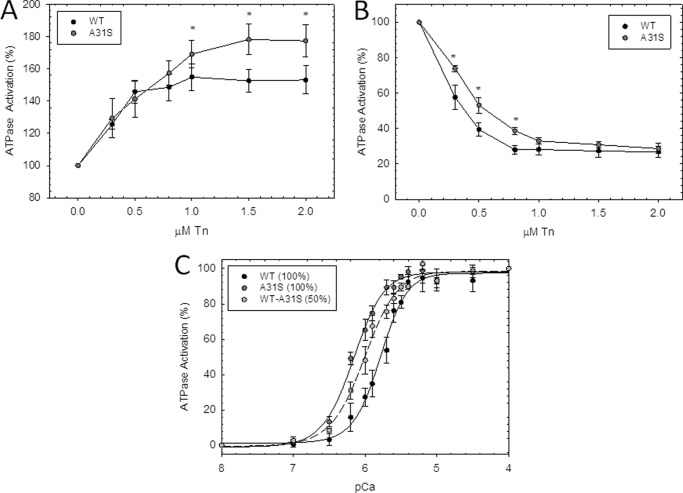
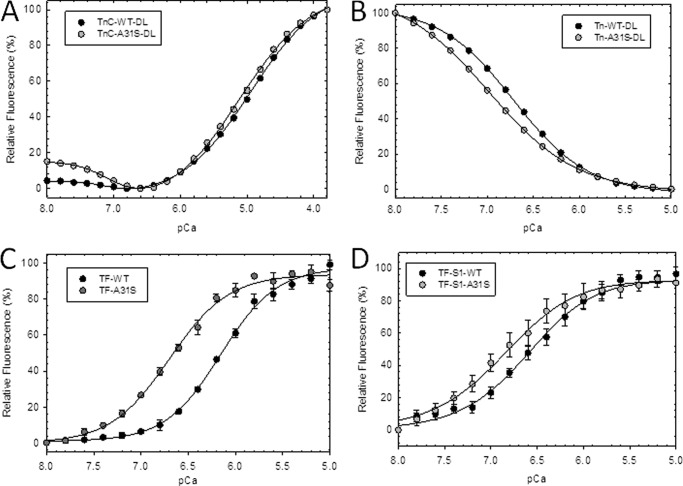
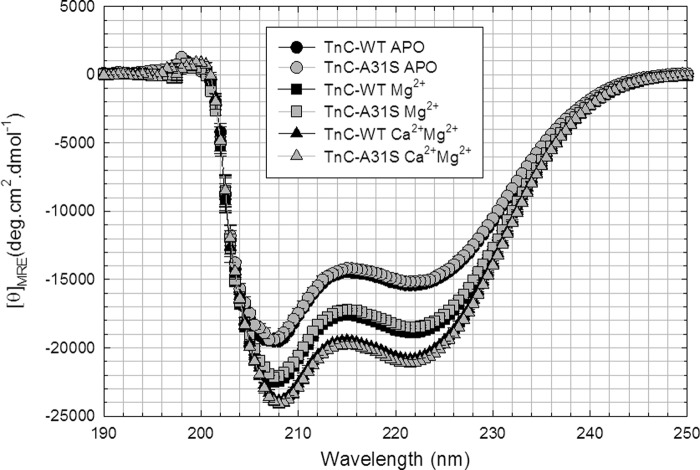
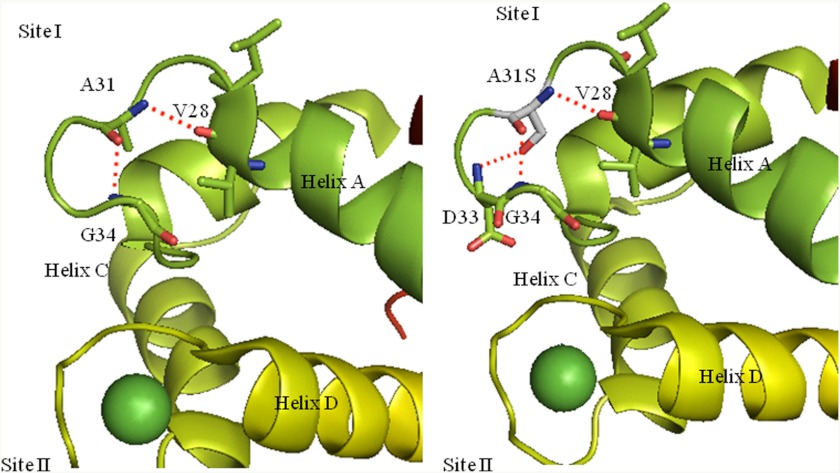
Defined as clinically unexplained hypertrophy of the left ventricle, hypertrophic cardiomyopathy (HCM) is traditionally understood as a disease of the cardiac sarcomere. Mutations in TNNC1-encoded cardiac troponin C (cTnC) are a relatively rare cause of HCM. Here, we report clinical and functional characterization of a novel TNNC1 mutation, A31S, identified in a pediatric HCM proband with multiple episodes of ventricular fibrillation and aborted sudden cardiac death. Diagnosed at age 5, the proband is family history-negative for HCM or sudden cardiac death, suggesting a de novo mutation. TnC-extracted cardiac skinned fibers were reconstituted with the cTnC-A31S mutant, which increased Ca(2+) sensitivity with no effect on the maximal contractile force generation. Reconstituted actomyosin ATPase assays with 50% cTnC-A31S:50% cTnC-WT demonstrated Ca(2+) sensitivity that was intermediate between 100% cTnC-A31S and 100% cTnC-WT, whereas the mutant increased the activation of the actomyosin ATPase without affecting the inhibitory qualities of the ATPase. The secondary structure of the cTnC mutant was evaluated by circular dichroism, which did not indicate global changes in structure. Fluorescence studies demonstrated increased Ca(2+) affinity in isolated cTnC, the troponin complex, thin filament, and to a lesser degree, thin filament with myosin subfragment 1. These results suggest that this mutation has a direct effect on the Ca(2+) sensitivity of the myofilament, which may alter Ca(2+) handling and contribute to the arrhythmogenesis observed in the proband. In summary, we report a novel mutation in the TNNC1 gene that is associated with HCM pathogenesis and may predispose to the pathogenesis of a fatal arrhythmogenic subtype of HCM.
Figures
References
-
- Maron B. J., Epstein S. E., Roberts W. C. (1986) Causes of sudden death in competitive athletes. J. Am. Coll. Cardiol. 7, 204–214 - PubMed
-
- Maron B. J., Gardin J. M., Flack J. M., Gidding S. S., Kurosaki T. T., Bild D. E. (1995) Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA study. Coronary artery risk development in (young) adults. Circulation 92, 785–789 - PubMed
-
- Maron B. J., Roberts W. C., McAllister H. A., Rosing D. R., Epstein S. E. (1980) Sudden death in young athletes. Circulation 62, 218–229 - PubMed
-
- Bos J. M., Towbin J. A., Ackerman M. J. (2009) Diagnostic, prognostic, and therapeutic implications of genetic testing for hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 54, 201–211 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
