

Myoclonus-dystonia syndrome due to tyrosine hydroxylase deficiency
- PMID: 22815559
- PMCID: PMC3405253
- DOI: 10.1212/WNL.0b013e318261714a
Myoclonus-dystonia syndrome due to tyrosine hydroxylase deficiency
Abstract
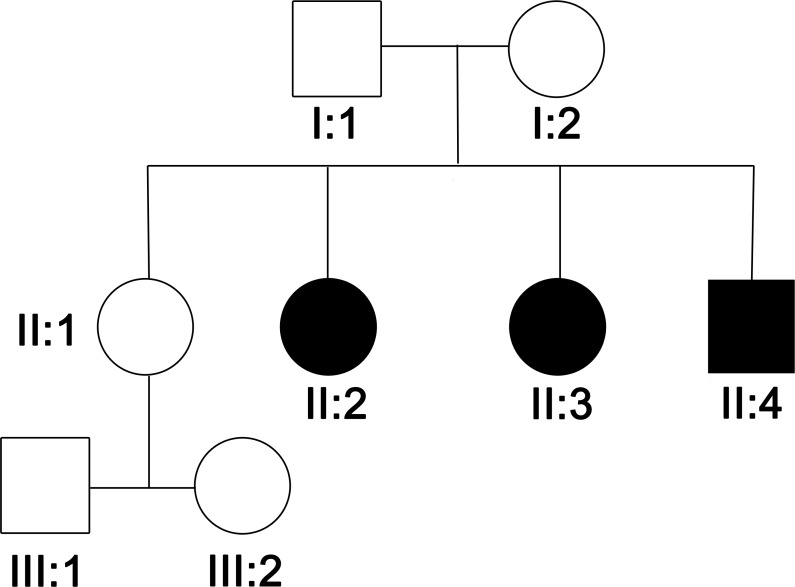
Objective: To present a new family with tyrosine hydroxylase deficiency (THD) that presented with a new phenotype of predominant, levodopa-responsive myoclonus with dystonia due to compound heterozygosity of one previously reported mutation in the promoter region and a novel nonsynonymous mutation in the other allele, thus expanding the clinical and genetic spectrum of this disorder.
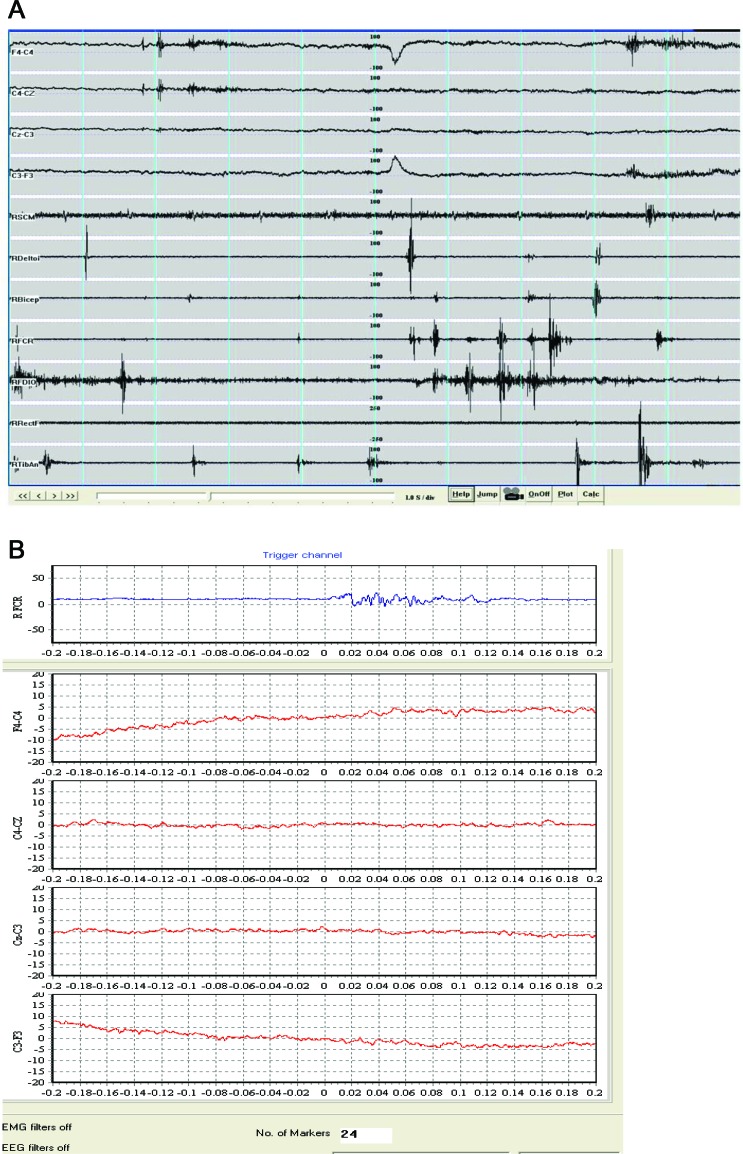
Methods: We performed detailed clinical examination of the family and electrophysiology to characterize the myoclonus. We performed analysis of the TH gene and in silico prediction of the possible effect of nonsynonymous substitutions on protein structure.
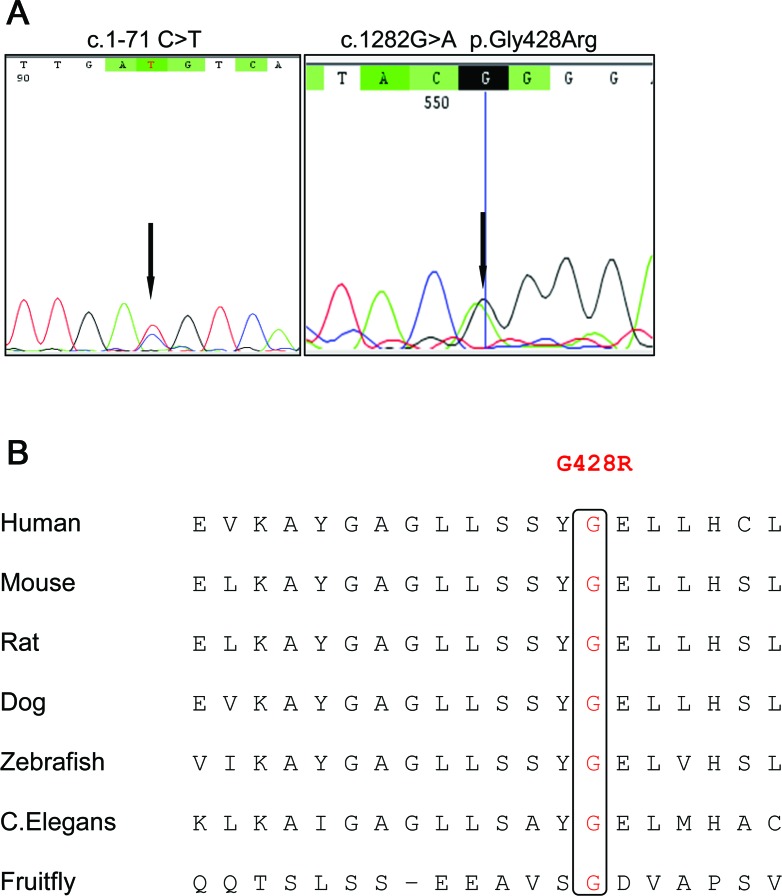
Results: Electrophysiology suggested that the myoclonus was of subcortical origin. Genetic analysis of the TH gene revealed compound heterozygosity of a point mutation in the promoter region (c.1-71 C>T) and a novel nonsynonymous substitution in exon 12 (c.1282G>A, p.Gly428Arg). The latter is a novel variant, predicted to have a deleterious effect on the TH protein function and is the first pathogenic TH mutation in patients of African ancestry.
Conclusion: We presented a THD family with predominant myoclonus-dystonia and a new genotype. It is important to consider THD in the differential diagnosis of myoclonus-dystonia, because early treatment with levodopa is crucial for these patients.
Figures
References
-
- Brautigam C, Wevers RA, Jansen RJT, et al. Biochemical hallmarks of tyrosine hydroxylase deficiency. Clin Chem 1998; 44: 1897– 1904 - PubMed
-
- Willemsen MA, Verbeek MM, Kamsteeg EJ, et al. Tyrosine hydroxylase deficiency: a treatable disorder of brain catecholamine biosynthesis. Brain 2010; 133: 1810– 1822 - PubMed
-
- Brautigam C, Steenbergen-Spanjers GCH, Hoffmann GF, et al. Biochemical and molecular genetic characteristics of the severe form of tyrosine hydroxylase deficiency. Clin Chem 1999; 45: 2073– 2078 - PubMed
-
- De Lonlay P, Nassogne MC, van Gennip AH, et al. Tyrosine hydroxylase deficiency unresponsive to L-dopa treatment with unusual clinical and biochemical presentation. J Inherit Metab Dis 2000; 23: 819– 825 - PubMed
-
- de Rijk-van Andel JF, Gabreels FJM, Geurtz B, et al. L-dopa-responsive infantile hypokinetic rigid parkinsonism due to tyrosine hydroxylase deficiency. Neurology 2000; 55: 1926– 1928 - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases