

Transcript dynamics of proinflammatory genes revealed by sequence analysis of subcellular RNA fractions
- PMID: 22817891
- PMCID: PMC3405548
- DOI: 10.1016/j.cell.2012.05.043
Transcript dynamics of proinflammatory genes revealed by sequence analysis of subcellular RNA fractions
Abstract
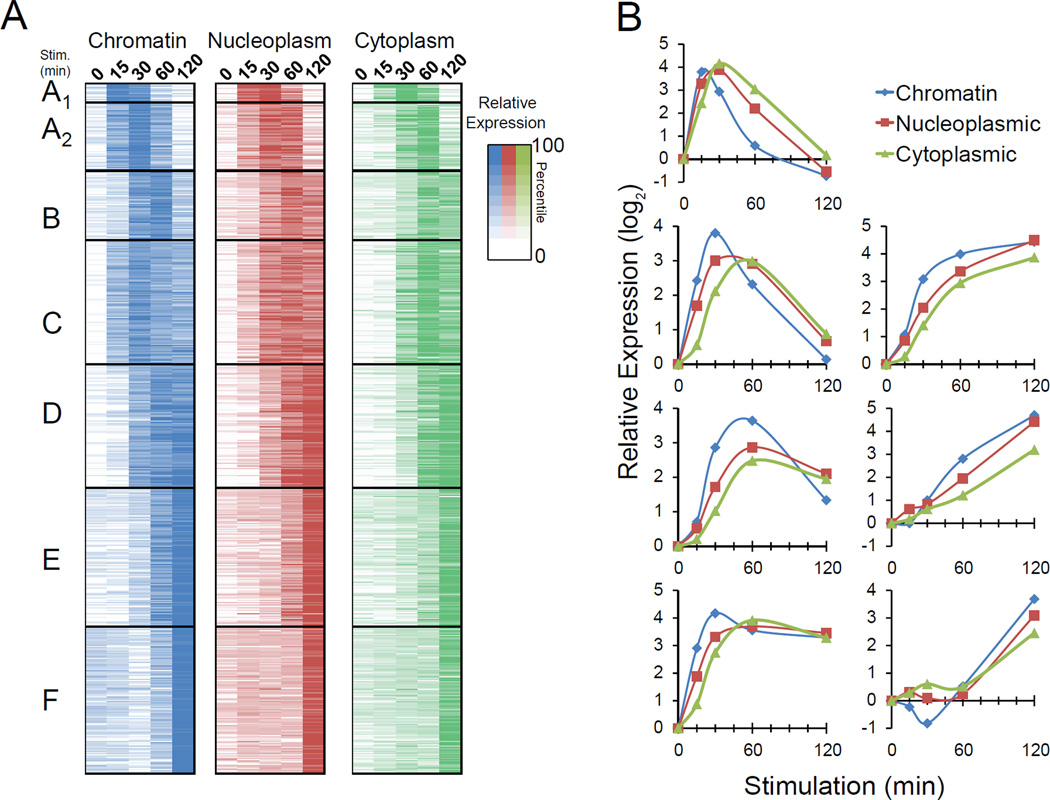
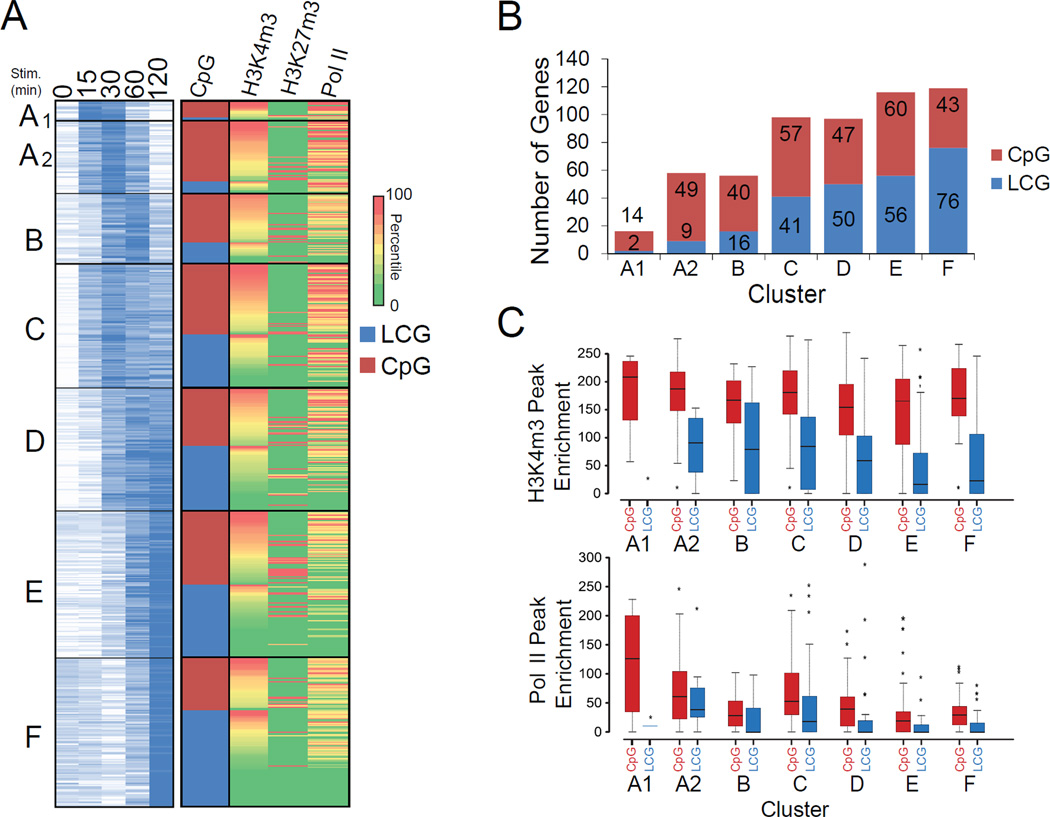
Macrophages respond to inflammatory stimuli by modulating the expression of hundreds of genes in a defined temporal cascade, with diverse transcriptional and posttranscriptional mechanisms contributing to the regulatory network. We examined proinflammatory gene regulation in activated macrophages by performing RNA-seq with fractionated chromatin-associated, nucleoplasmic, and cytoplasmic transcripts. This methodological approach allowed us to separate the synthesis of nascent transcripts from transcript processing and the accumulation of mature mRNAs. In addition to documenting the subcellular locations of coding and noncoding transcripts, the results provide a high-resolution view of the relationship between defined promoter and chromatin properties and the temporal regulation of diverse classes of coexpressed genes. The data also reveal a striking accumulation of full-length yet incompletely spliced transcripts in the chromatin fraction, suggesting that splicing often occurs after transcription has been completed, with transcripts retained on the chromatin until fully spliced.
Copyright © 2012 Elsevier Inc. All rights reserved.
Figures





Comment in
-
Transcription, splicing, and release: are we there yet?Cell. 2012 Jul 20;150(2):241-3. doi: 10.1016/j.cell.2012.07.004. Cell. 2012. PMID: 22817885
-
Splicing: waiting to be spliced.Nat Rev Genet. 2012 Sep;13(9):599. doi: 10.1038/nrg3310. Epub 2012 Aug 7. Nat Rev Genet. 2012. PMID: 22868265 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
