

Pre-mRNA splicing in disease and therapeutics
- PMID: 22819011
- PMCID: PMC3411911
- DOI: 10.1016/j.molmed.2012.06.006
Pre-mRNA splicing in disease and therapeutics
Abstract
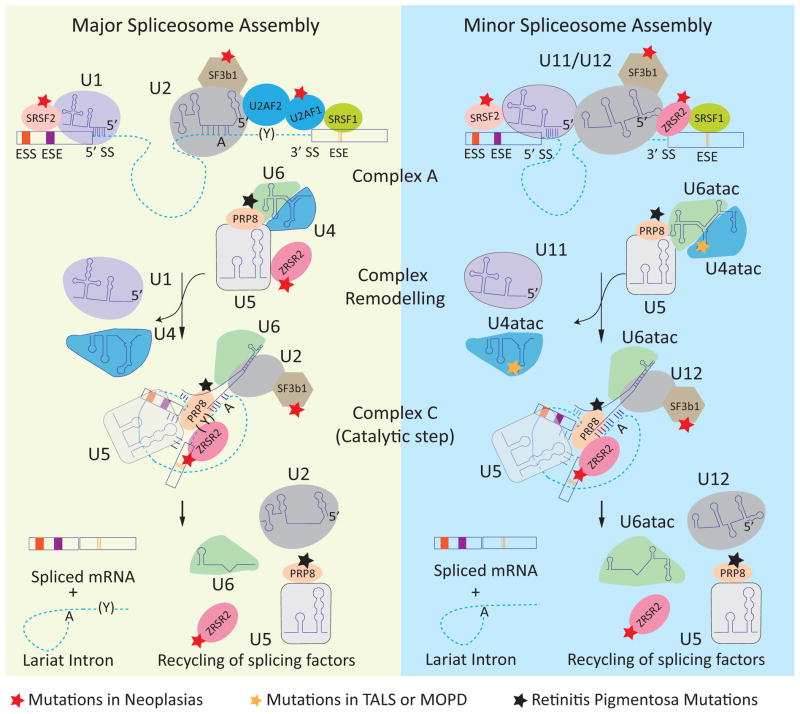
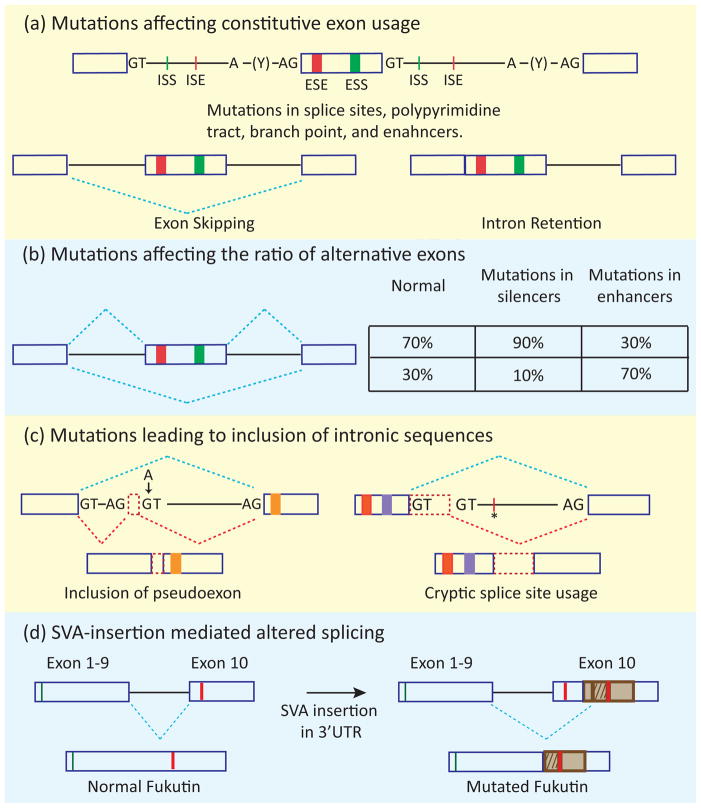
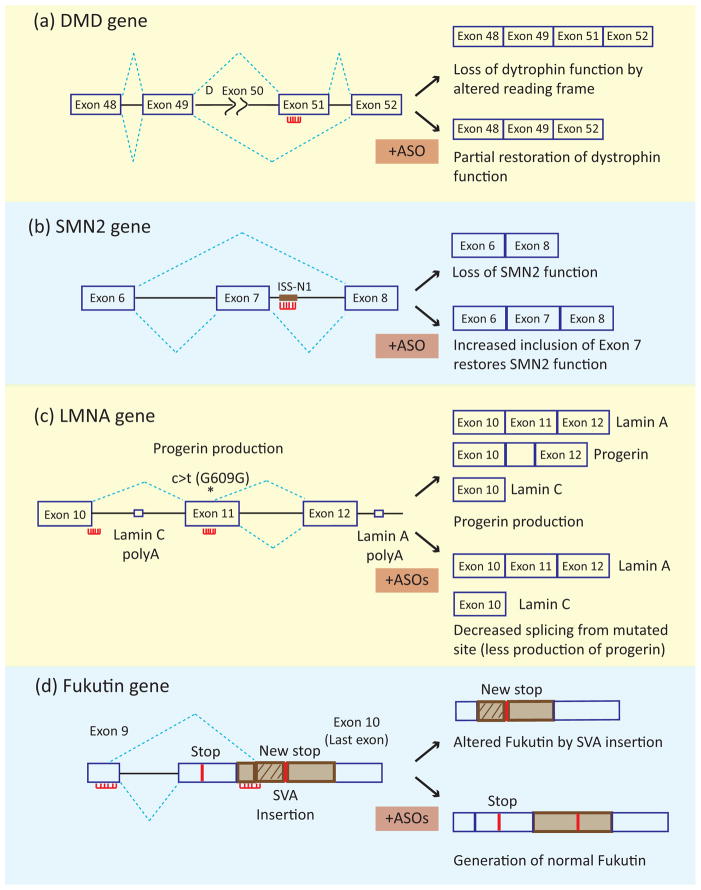
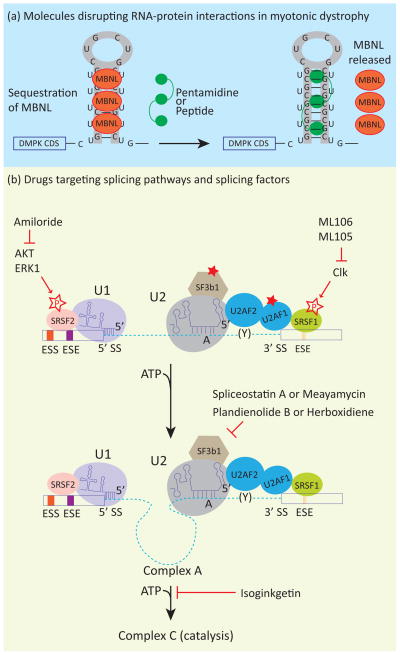
In metazoans, alternative splicing of genes is essential for regulating gene expression and contributing to functional complexity. Computational predictions, comparative genomics, and transcriptome profiling of normal and diseased tissues indicate that an unexpectedly high fraction of diseases are caused by mutations that alter splicing. Mutations in cis elements cause missplicing of genes that alter gene function and contribute to disease pathology. Mutations of core spliceosomal factors are associated with hematolymphoid neoplasias, retinitis pigmentosa, and microcephalic osteodysplastic primordial dwarfism type 1 (MOPD1). Mutations in the trans regulatory factors that control alternative splicing are associated with autism spectrum disorder, amyotrophic lateral sclerosis (ALS), and various cancers. In addition to discussing the disorders caused by these mutations, this review summarizes therapeutic approaches that have emerged to correct splicing of individual genes or target the splicing machinery.
Copyright © 2012 Elsevier Ltd. All rights reserved.
Figures
References
-
- Wahl MC, et al. The spliceosome: design principles of a dynamic RNP machine. Cell. 2009;136:701–718. - PubMed
-
- Berget SM. Exon recognition in vertebrate splicing. J Biol Chem. 1995;270:2411–2414. - PubMed
-
- Patel AA, Steitz JA. Splicing double: insights from the second spliceosome. Nat Rev Mol Cell Biol. 2003;4:960–970. - PubMed
-
- Will CL, Luhrmann R. Splicing of a rare class of introns by the U12-dependent spliceosome. Biol Chem. 2005;386:713–724. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
