

Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells
- PMID: 22820318
- PMCID: PMC3467340
- DOI: 10.1038/nbt.2282
Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells
Erratum in
-
Author Correction: Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells.Nat Biotechnol. 2020 Mar;38(3):374. doi: 10.1038/s41587-020-0427-1. Nat Biotechnol. 2020. PMID: 32015550
Abstract
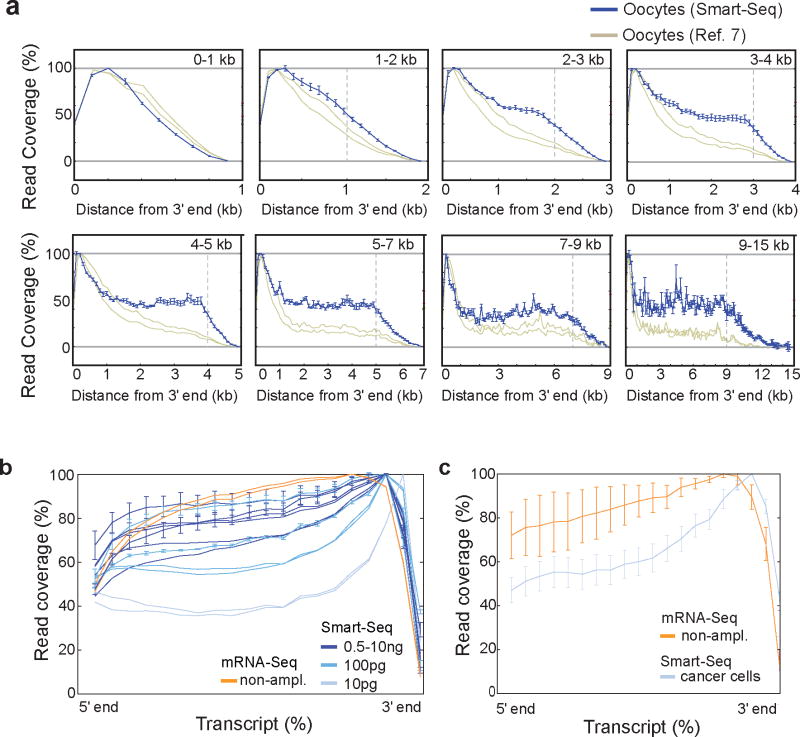
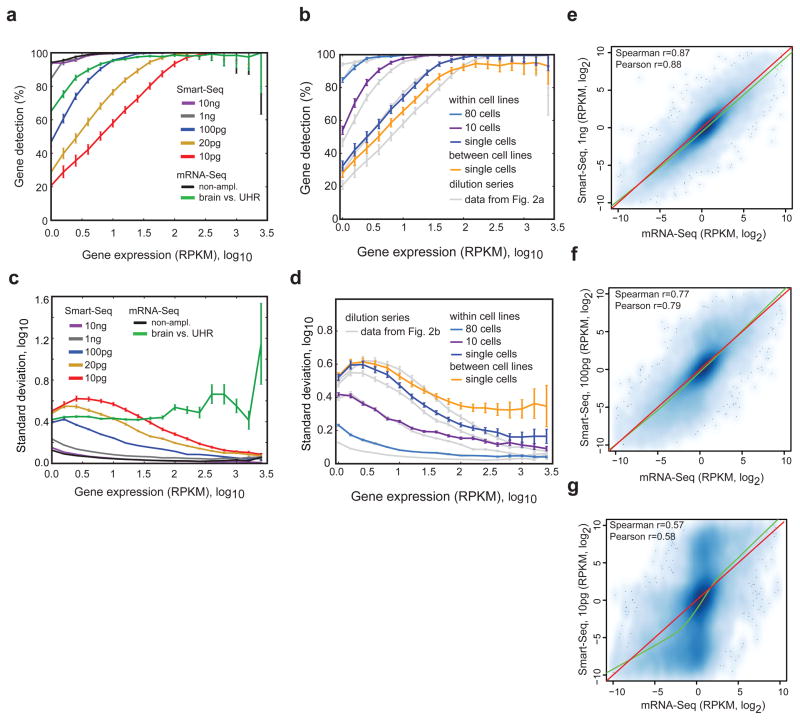
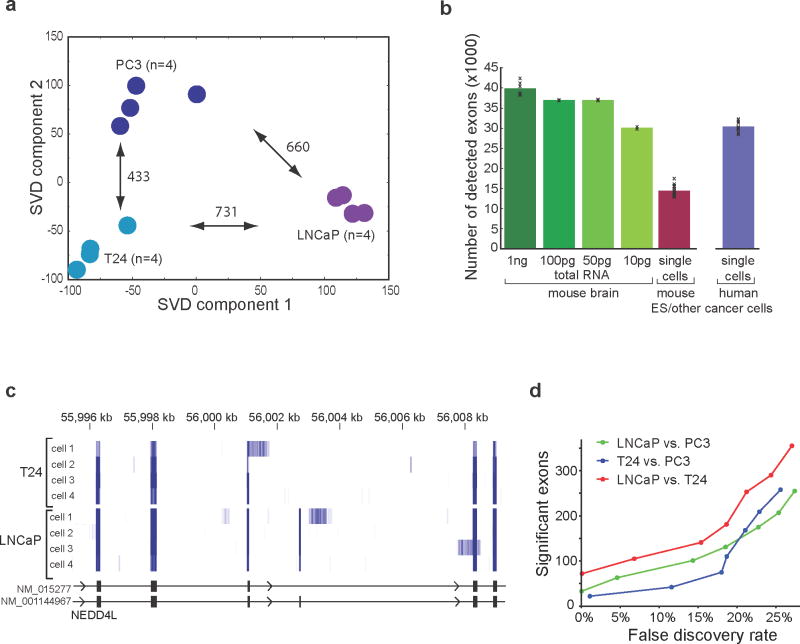
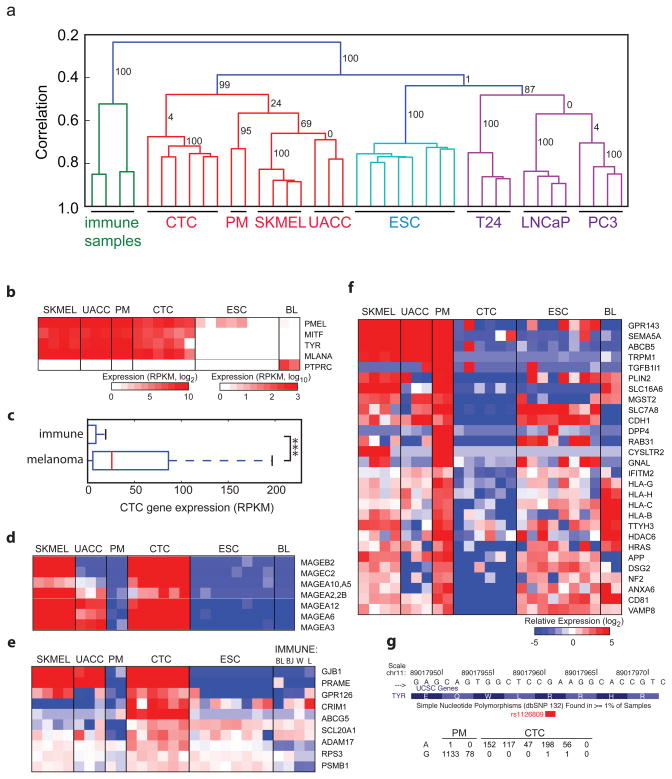
Genome-wide transcriptome analyses are routinely used to monitor tissue-, disease- and cell type–specific gene expression, but it has been technically challenging to generate expression profiles from single cells. Here we describe a robust mRNA-Seq protocol (Smart-Seq) that is applicable down to single cell levels. Compared with existing methods, Smart-Seq has improved read coverage across transcripts, which enhances detailed analyses of alternative transcript isoforms and identification of single-nucleotide polymorphisms. We determined the sensitivity and quantitative accuracy of Smart-Seq for single-cell transcriptomics by evaluating it on total RNA dilution series. We found that although gene expression estimates from single cells have increased noise, hundreds of differentially expressed genes could be identified using few cells per cell type. Applying Smart-Seq to circulating tumor cells from melanomas, we identified distinct gene expression patterns, including candidate biomarkers for melanoma circulating tumor cells. Our protocol will be useful for addressing fundamental biological problems requiring genome-wide transcriptome profiling in rare cells.
Conflict of interest statement
The reported sequence read data have been deposited in Gene Expression Omnibus at NCBI (GSE38495). S.L., R.L., I.K., and G.P.S. are employees and shareholders of Illumina Inc, but the remaining authors have no competing financial interests.
Figures
Comment in
-
Transcriptome sequencing of single cells with Smart-Seq.Nat Biotechnol. 2012 Aug;30(8):763-5. doi: 10.1038/nbt.2325. Nat Biotechnol. 2012. PMID: 22871714 No abstract available.
References
-
- Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nature Genetics. 2008;40:1413–1415. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
