

Current challenges in software solutions for mass spectrometry-based quantitative proteomics
- PMID: 22821268
- PMCID: PMC3418498
- DOI: 10.1007/s00726-012-1289-8
Current challenges in software solutions for mass spectrometry-based quantitative proteomics
Abstract
Mass spectrometry-based proteomics has evolved as a high-throughput research field over the past decade. Significant advances in instrumentation, and the ability to produce huge volumes of data, have emphasized the need for adequate data analysis tools, which are nowadays often considered the main bottleneck for proteomics development. This review highlights important issues that directly impact the effectiveness of proteomic quantitation and educates software developers and end-users on available computational solutions to correct for the occurrence of these factors. Potential sources of errors specific for stable isotope-based methods or label-free approaches are explicitly outlined. The overall aim focuses on a generic proteomic workflow.
Figures
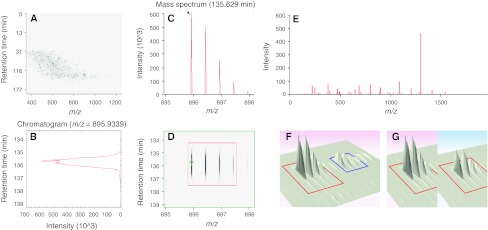
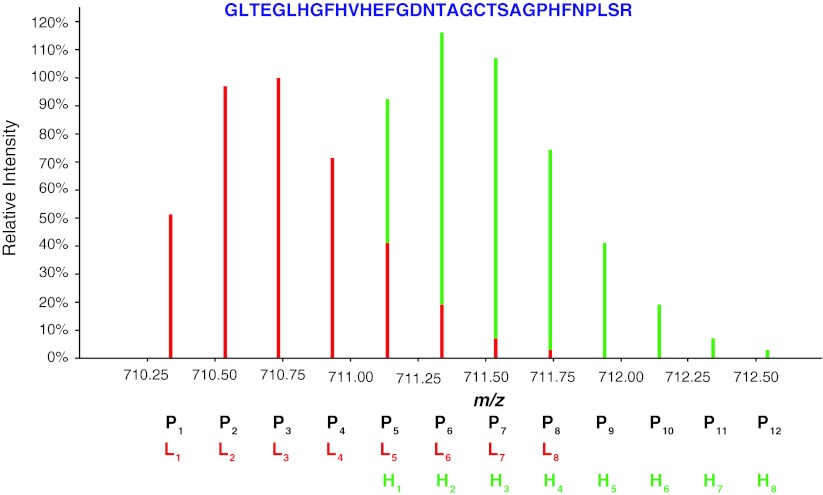
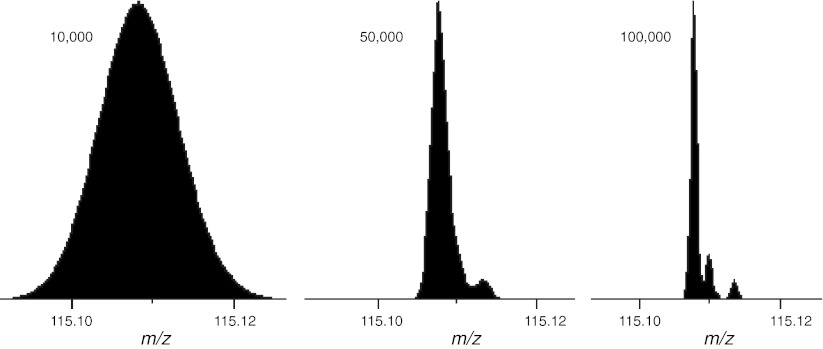
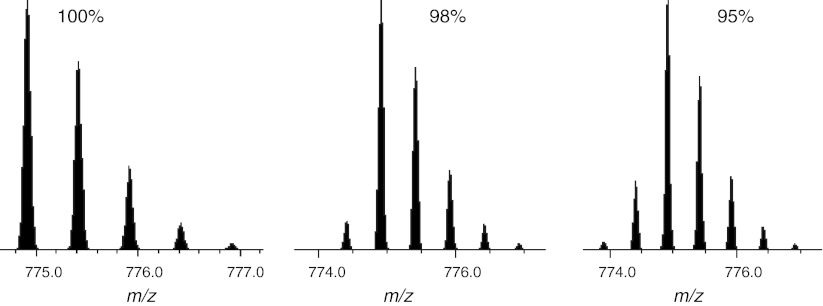
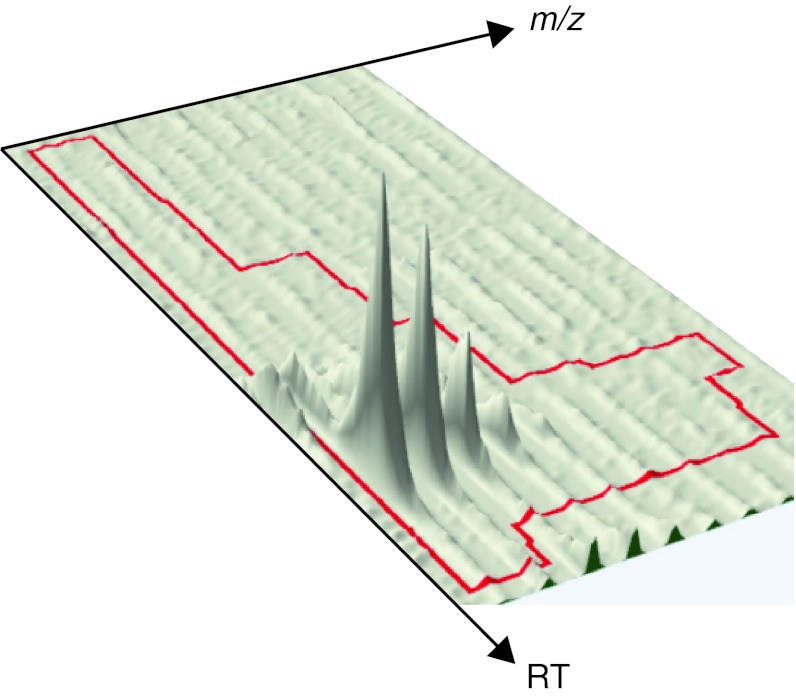
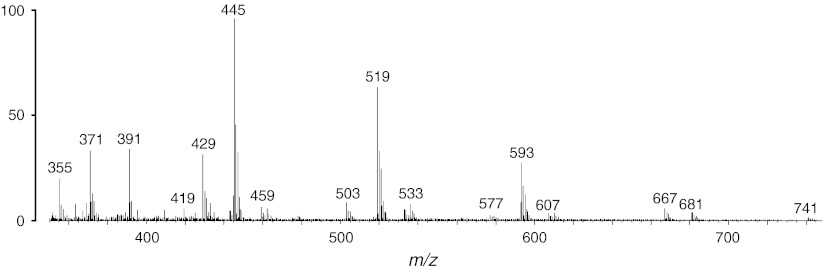
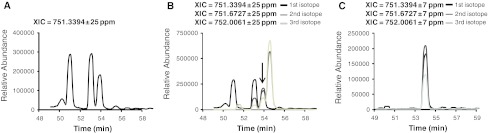
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
