

Myeloid malignancies: mutations, models and management
- PMID: 22823977
- PMCID: PMC3418560
- DOI: 10.1186/1471-2407-12-304
Myeloid malignancies: mutations, models and management
Abstract
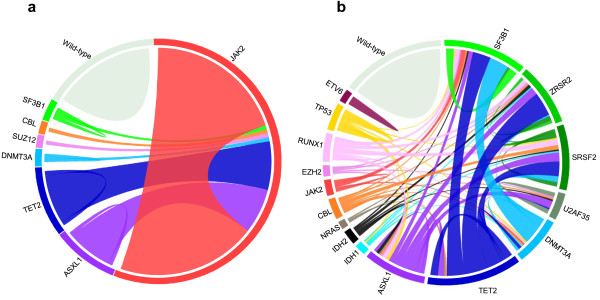
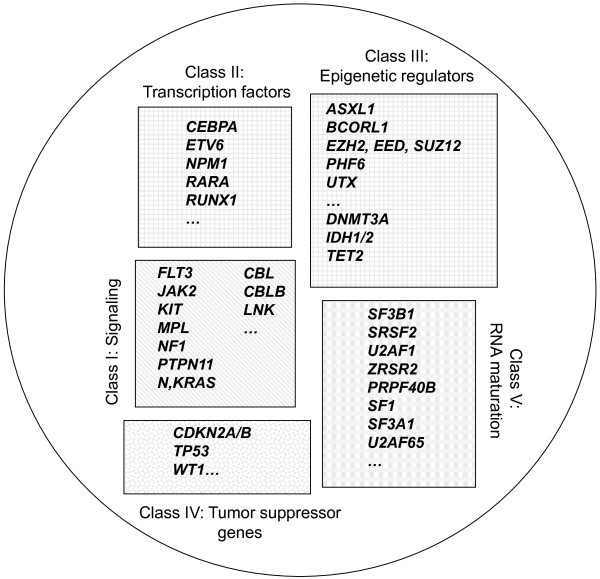
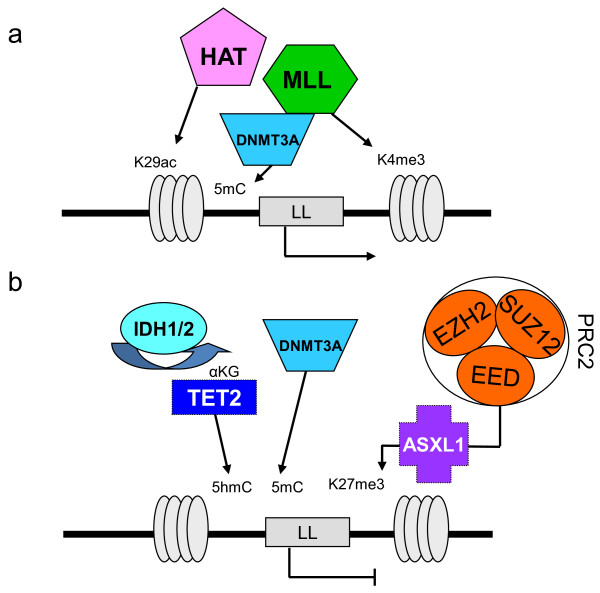
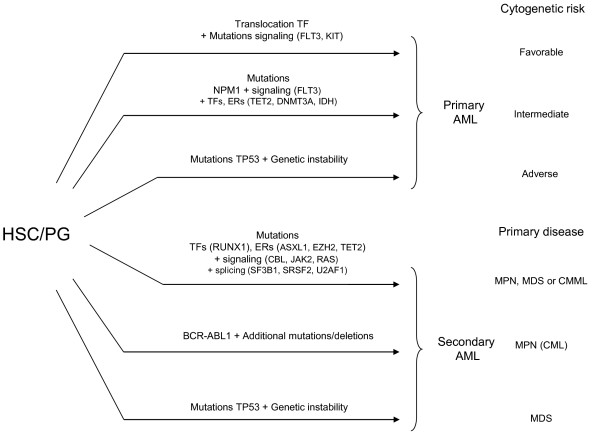
Myeloid malignant diseases comprise chronic (including myelodysplastic syndromes, myeloproliferative neoplasms and chronic myelomonocytic leukemia) and acute (acute myeloid leukemia) stages. They are clonal diseases arising in hematopoietic stem or progenitor cells. Mutations responsible for these diseases occur in several genes whose encoded proteins belong principally to five classes: signaling pathways proteins (e.g. CBL, FLT3, JAK2, RAS), transcription factors (e.g. CEBPA, ETV6, RUNX1), epigenetic regulators (e.g. ASXL1, DNMT3A, EZH2, IDH1, IDH2, SUZ12, TET2, UTX), tumor suppressors (e.g. TP53), and components of the spliceosome (e.g. SF3B1, SRSF2). Large-scale sequencing efforts will soon lead to the establishment of a comprehensive repertoire of these mutations, allowing for a better definition and classification of myeloid malignancies, the identification of new prognostic markers and therapeutic targets, and the development of novel therapies. Given the importance of epigenetic deregulation in myeloid diseases, the use of drugs targeting epigenetic regulators appears as a most promising therapeutic approach.
Figures
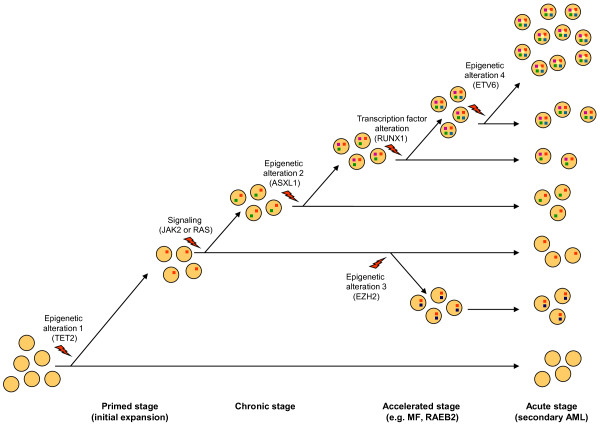
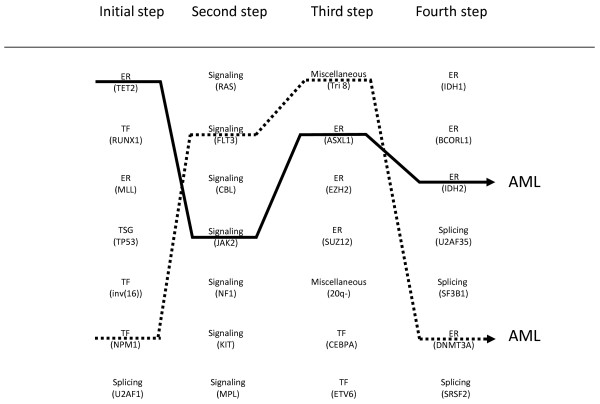
References
-
- Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, Vassiliou GS, Bench AJ, Boyd EM, Curtin N, Scott MA, Erber WN, Green AR. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365:1054–1061. - PubMed
-
- James C, Ugo V, Le Couedic JP, Staerk J, Delhommeau F, Lacout C, Garcon L, Raslova H, Berger R, Bennaceur-Griscelli A, Villeval JL, Constantinescu SN, Casadevall N, Vainchenker W. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434:1144–1148. - PubMed
-
- Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, Tichelli A, Cazzola M, Skoda RC. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352:1779–1790. - PubMed
-
- Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, Boggon TJ, Wlodarska I, Clark JJ, Moore S, Adelsperger J, Koo S, Lee JC, Gabriel S, Mercher T, D'Andrea A, Frohling S, Dohner K, Marynen P, Vandenberghe P, Mesa RA, Tefferi A, Griffin JD, Eck MJ, Sellers WR, Meyerson M, Golub TR, Lee SJ, Gilliland DG. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7:387–397. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
