

Swept field laser confocal microscopy for enhanced spatial and temporal resolution in live-cell imaging
- PMID: 22831554
- PMCID: PMC3549604
- DOI: 10.1017/S1431927612000542
Swept field laser confocal microscopy for enhanced spatial and temporal resolution in live-cell imaging
Abstract
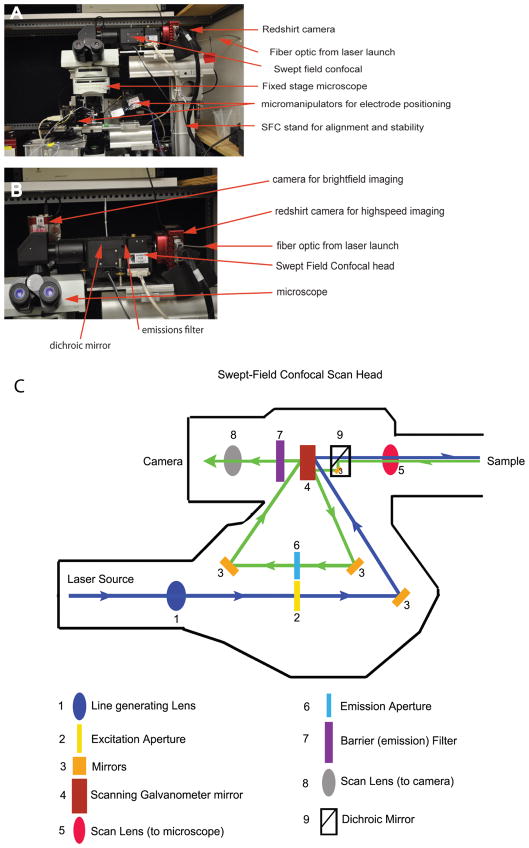
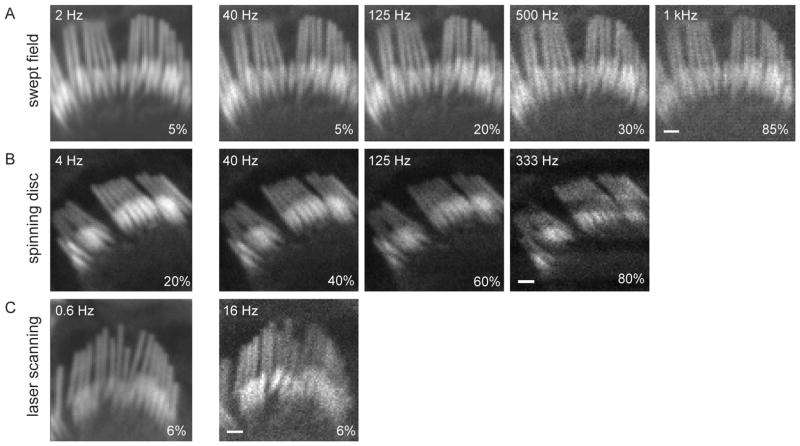
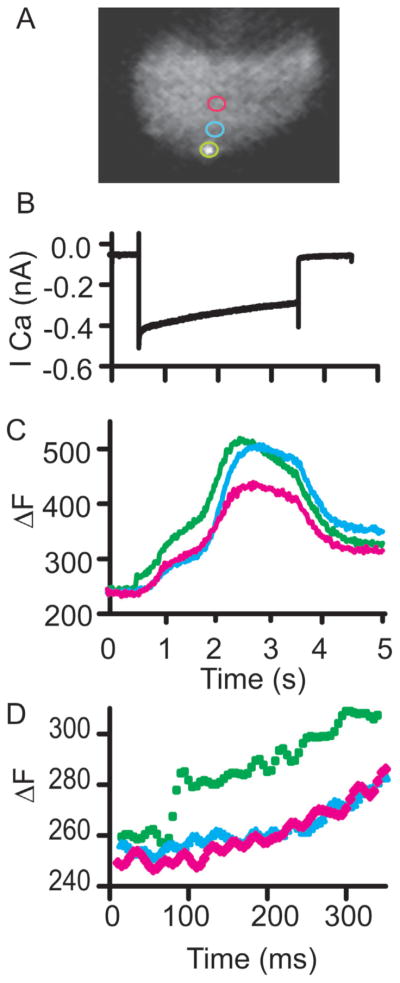
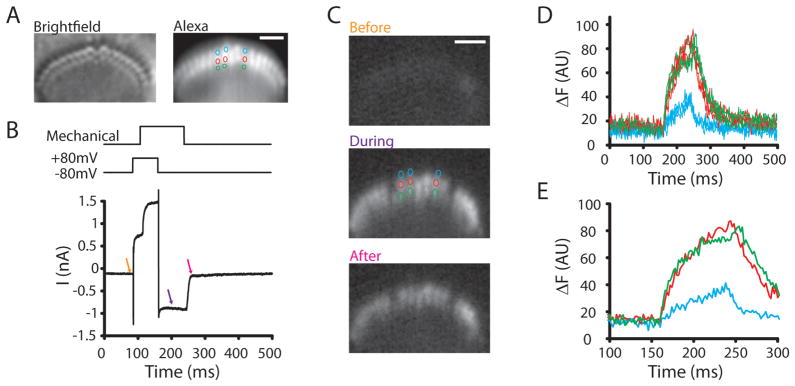
Confocal fluorescence microscopy is a broadly used imaging technique that enhances the signal-to-noise ratio by removing out of focal plane fluorescence. Confocal microscopes come with a variety of modifications depending on the particular experimental goals. Microscopes, illumination pathways, and light collection were originally focused upon obtaining the highest resolution image possible, typically on fixed tissue. More recently, live-cell confocal imaging has gained importance. Since measured signals are often rapid or transient, thus requiring higher sampling rates, specializations are included to enhance spatial and temporal resolution while maintaining tissue viability. Thus, a balance between image quality, temporal resolution, and tissue viability is needed. A subtype of confocal imaging, termed swept field confocal (SFC) microscopy, can image live cells at high rates while maintaining confocality. SFC systems can use a pinhole array to obtain high spatial resolution, similar to spinning disc systems. In addition, SFC imaging can achieve faster rates by using a slit to sweep the light across the entire image plane, thus requiring a single scan to generate an image. Coupled to a high-speed charge-coupled device camera and a laser illumination source, images can be obtained at greater than 1,000 frames per second while maintaining confocality.
Figures


References
-
- Gratton E, vandeVen MJ. Chapter 5: Laser sources for confocal microscopy. In: Pawley JB, editor. Handbook of Biological Confocal Microscopy. 3. New York: Springer; 2006. pp. 80–125.
-
- Inoue S. Chapter 1: Foundations of confocal scanned imaging in light microscopy. In: Pawley JB, editor. Handbook of Biological Confocal Microscopy. 3. New York: Springer; 2006. pp. 1–16.
-
- Minsky M. Memoir on inventing the confocal microscope. Scanning. 1988;10:128–138.
