

Mechanisms underlying the rapid peroxisome proliferator-activated receptor-γ-mediated amyloid clearance and reversal of cognitive deficits in a murine model of Alzheimer's disease
- PMID: 22836247
- PMCID: PMC3433721
- DOI: 10.1523/JNEUROSCI.5268-11.2012
Mechanisms underlying the rapid peroxisome proliferator-activated receptor-γ-mediated amyloid clearance and reversal of cognitive deficits in a murine model of Alzheimer's disease
Abstract
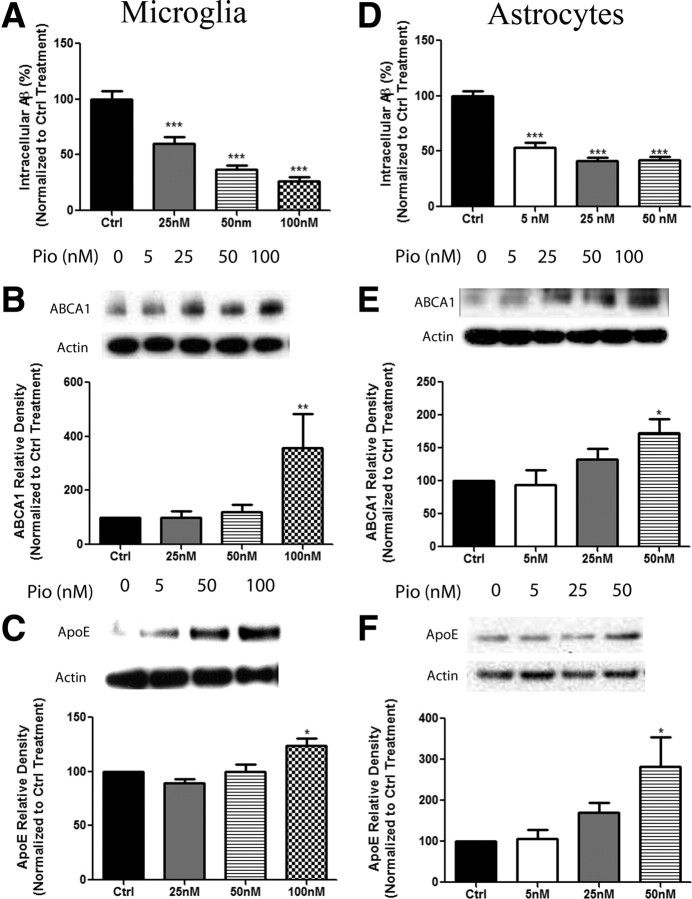
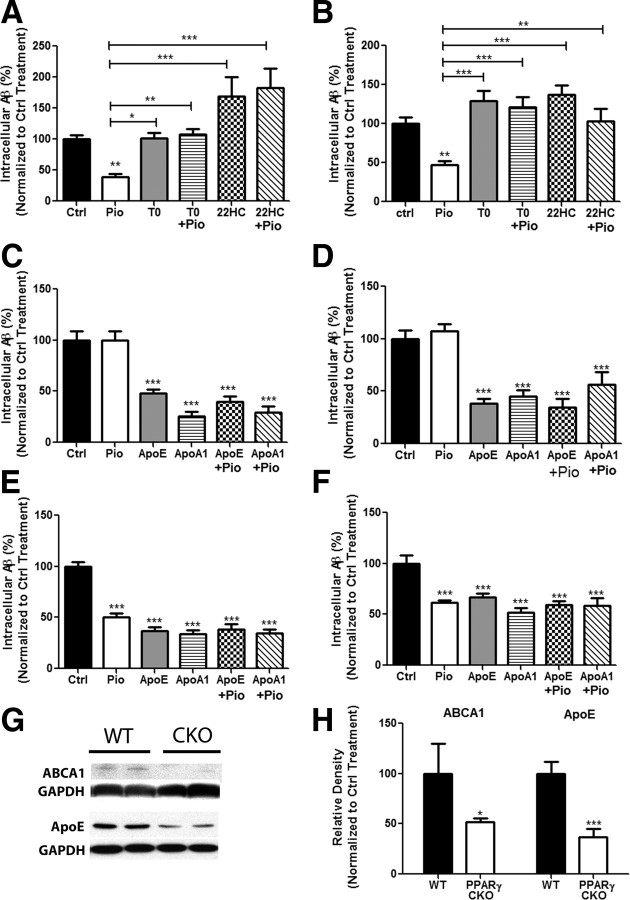
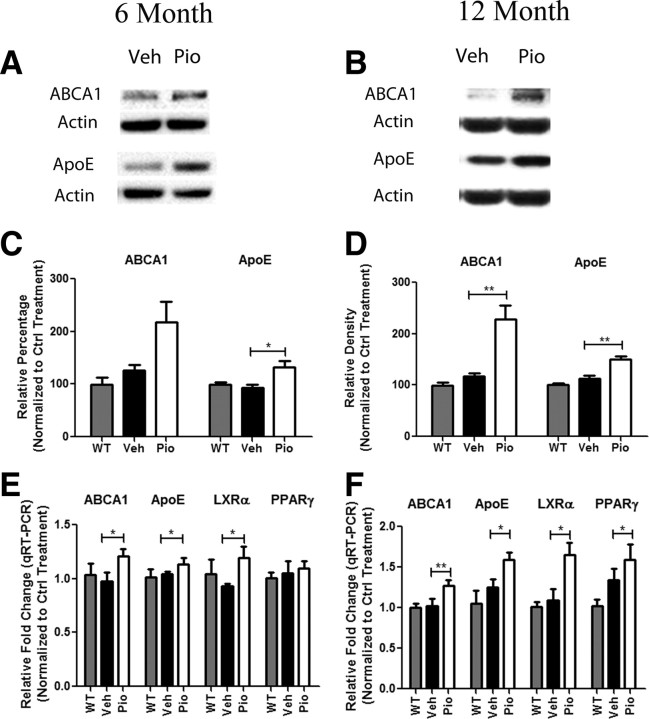
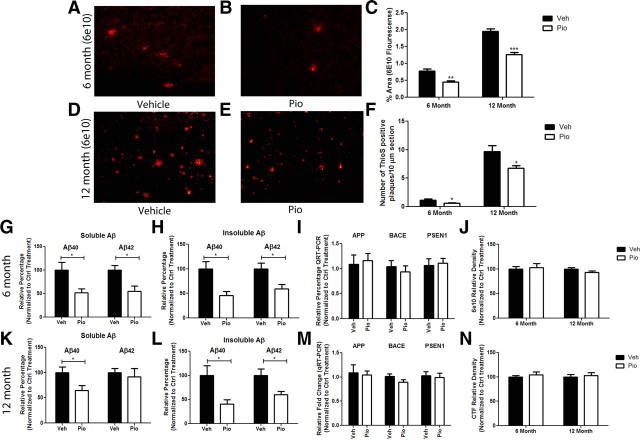
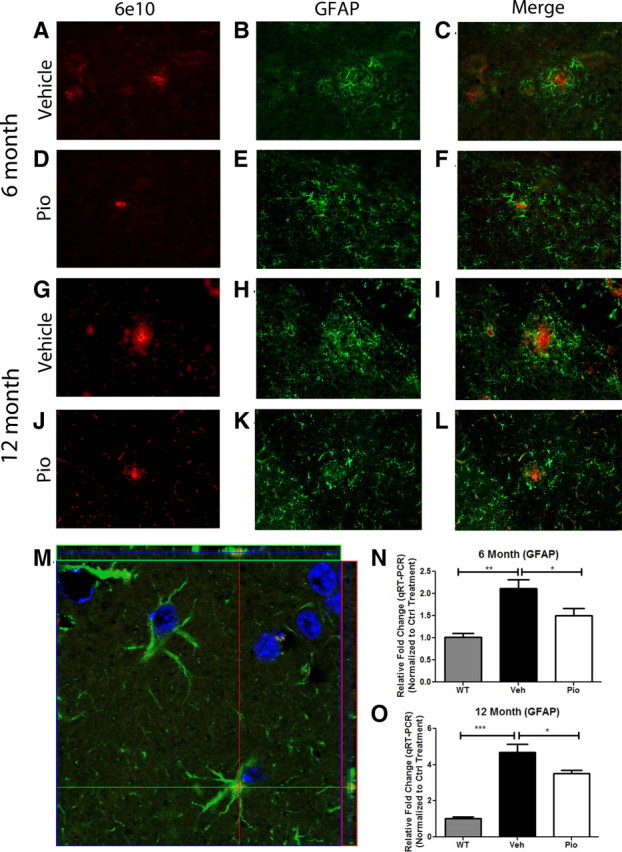
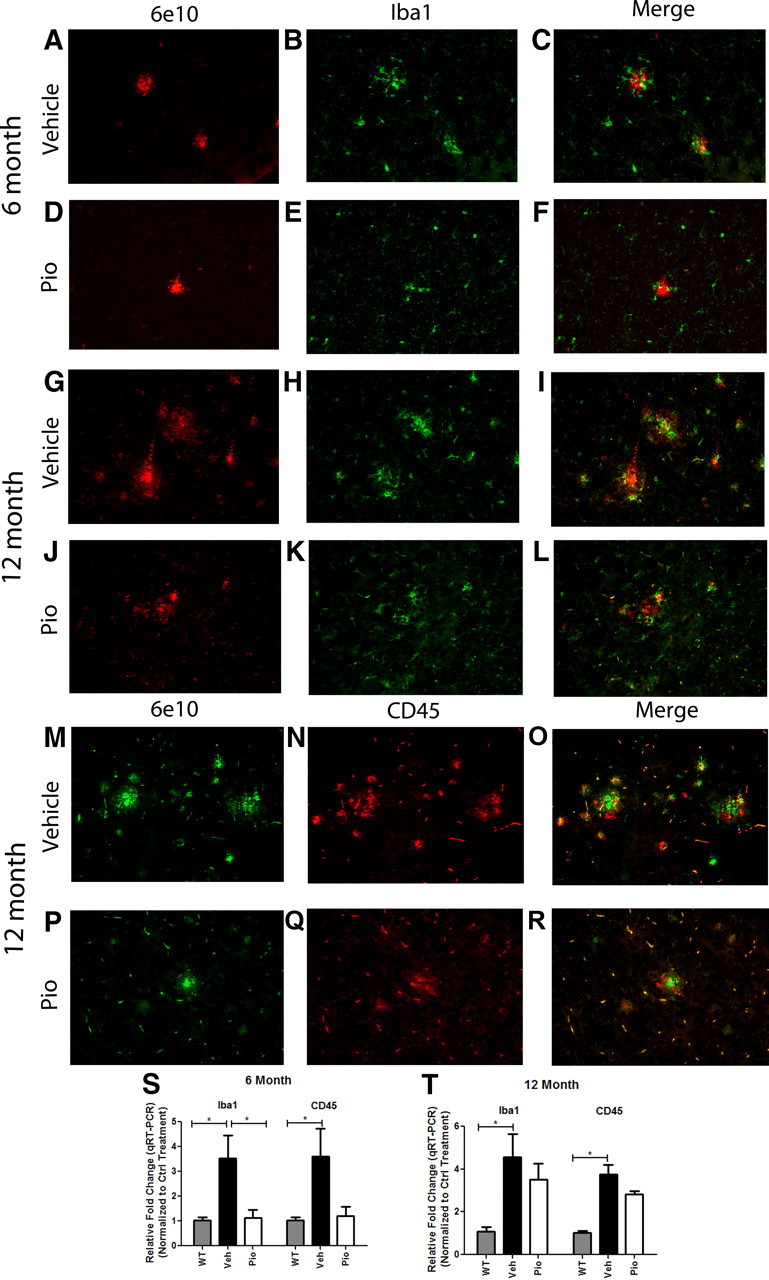
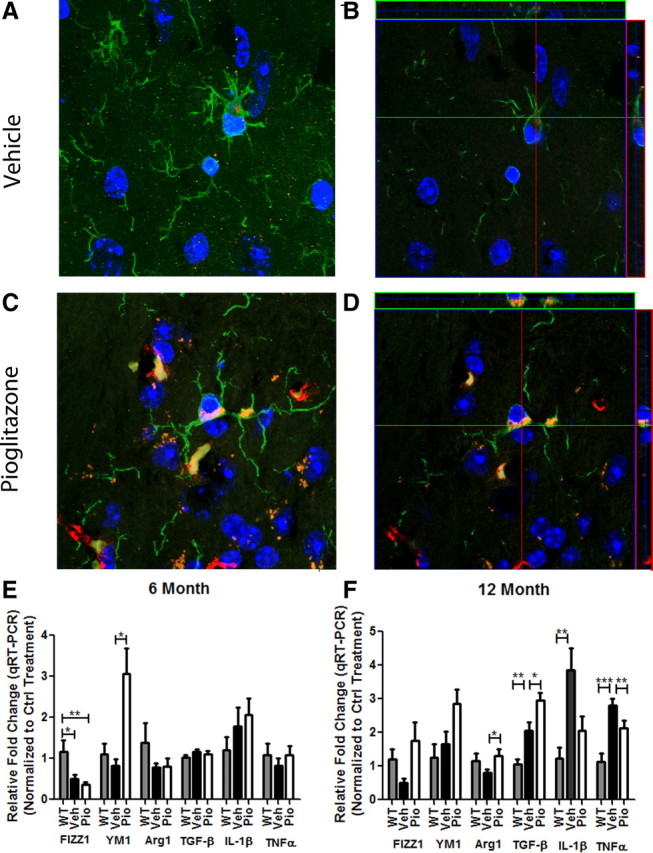
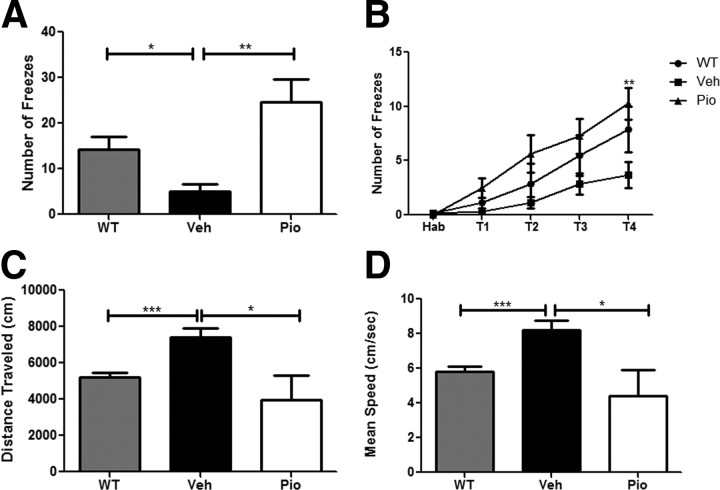
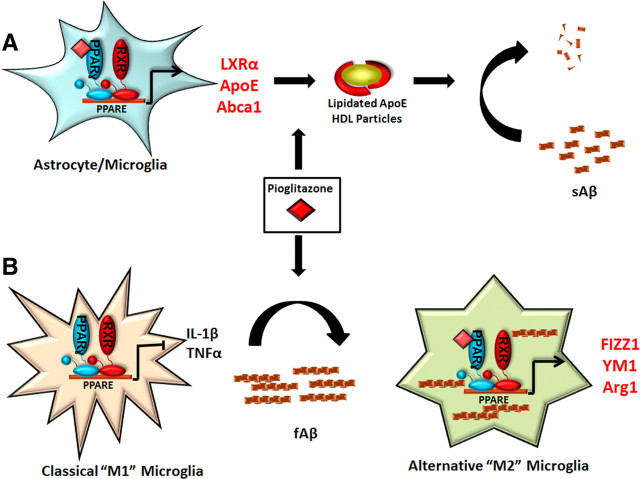
Alzheimer's disease is associated with a disruption of amyloid β (Aβ) homeostasis, resulting in the accumulation and subsequent deposition of Aβ peptides within the brain. The peroxisome proliferator-activated receptor-γ (PPARγ) is a ligand-activated nuclear receptor that acts in a coupled metabolic cycle with Liver X Receptors (LXRs) to increase brain apolipoprotein E (apoE) levels. apoE functions to promote the proteolytic clearance of soluble forms of Aβ, and we found that the synthetic PPARγ agonist, pioglitazone, stimulated Aβ degradation by both microglia and astrocytes in an LXR and apoE-dependent manner. Remarkably, a brief 9 d oral treatment of APPswe/PS1Δe9 mice with pioglitazone resulted in dramatic reductions in brain levels of soluble and insoluble Aβ levels which correlated with the loss of both diffuse and dense-core plaques within the cortex. The removal of preexisting amyloid deposits was associated with the appearance of abundant Aβ-laden microglia and astrocytes. Pioglitazone treatment resulted in the phenotypic polarization of microglial cells from a proinflammatory M1 state, into an anti-inflammatory M2 state that was associated with enhanced phagocytosis of deposited forms of amyloid. The reduction in amyloid levels was associated with a reversal of contextual memory deficits in the drug-treated mice. These data provide a mechanistic explanation for how PPARγ activation facilitates amyloid clearance and supports the therapeutic utility of PPARγ agonists for the treatment of Alzheimer's disease.
Figures
References
-
- Akiyama H, Arai T, Kondo H, Tanno E, Haga C, Ikeda K. Cell mediators of inflammation in the Alzheimer disease brain. Alzheimer Dis Assoc Disord. 2000a;14(Suppl 1):S47–S53. - PubMed
-
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, et al. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000b;21:383–421. - PMC - PubMed
-
- Beaven SW, Tontonoz P. Nuclear receptors in lipid metabolism: targeting the heart of dyslipidemia. Annu Rev Med. 2006;57:313–329. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous