

Dissecting DNA damage response pathways by analysing protein localization and abundance changes during DNA replication stress
- PMID: 22842922
- PMCID: PMC3434236
- DOI: 10.1038/ncb2549
Dissecting DNA damage response pathways by analysing protein localization and abundance changes during DNA replication stress
Abstract
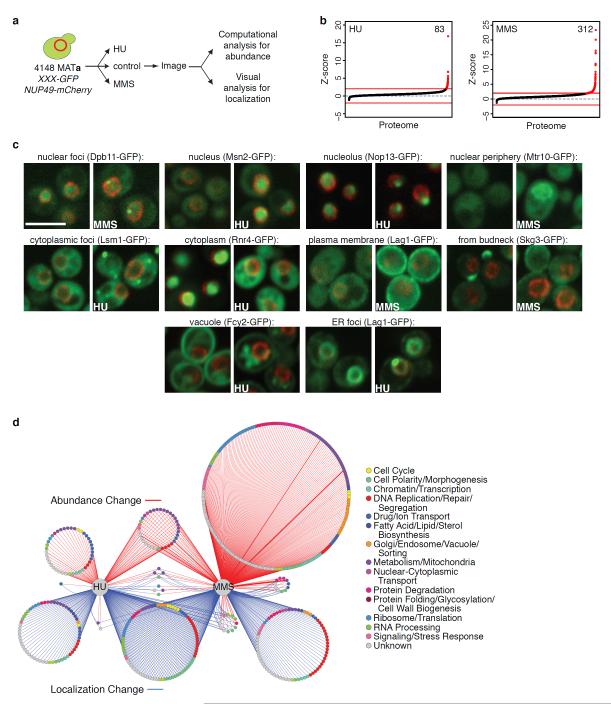
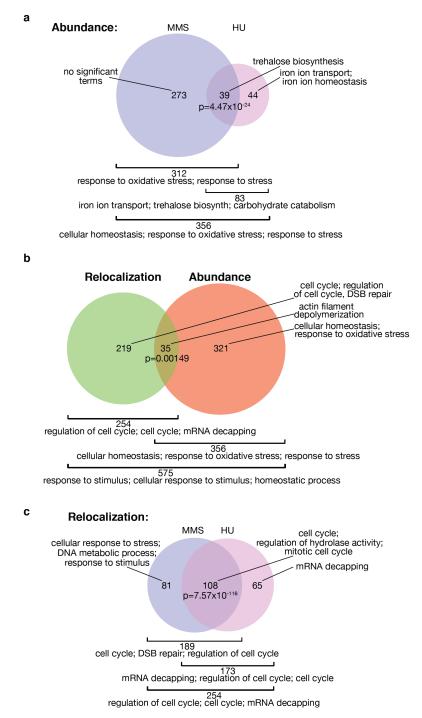
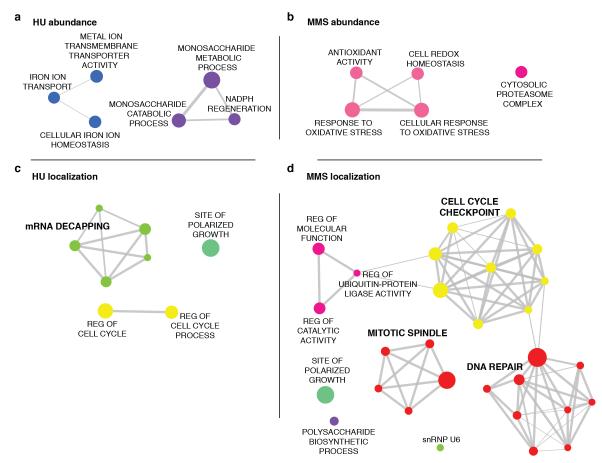
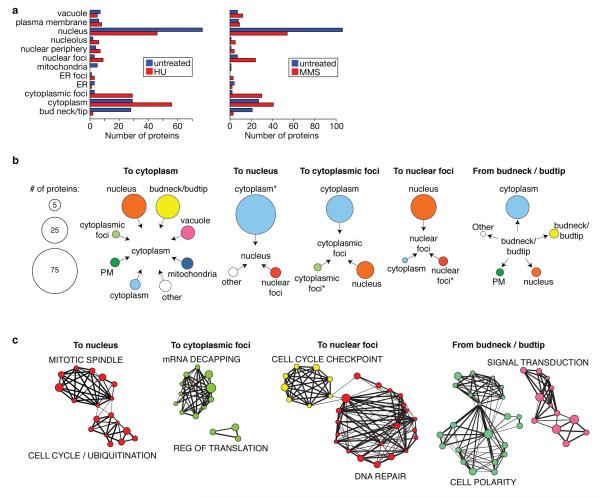
Relocalization of proteins is a hallmark of the DNA damage response. We use high-throughput microscopic screening of the yeast GFP fusion collection to develop a systems-level view of protein reorganization following drug-induced DNA replication stress. Changes in protein localization and abundance reveal drug-specific patterns of functional enrichments. Classification of proteins by subcellular destination enables the identification of pathways that respond to replication stress. We analysed pairwise combinations of GFP fusions and gene deletion mutants to define and order two previously unknown DNA damage responses. In the first, Cmr1 forms subnuclear foci that are regulated by the histone deacetylase Hos2 and are distinct from the typical Rad52 repair foci. In a second example, we find that the checkpoint kinases Mec1/Tel1 and the translation regulator Asc1 regulate P-body formation. This method identifies response pathways that were not detected in genetic and protein interaction screens, and can be readily applied to any form of chemical or genetic stress to reveal cellular response pathways.
Figures
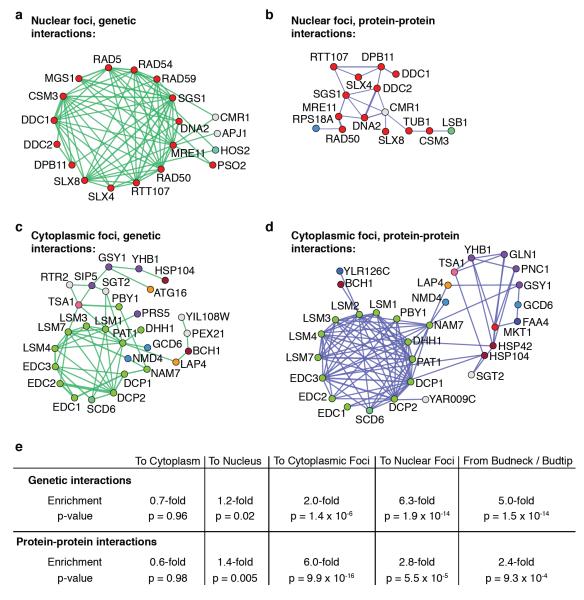
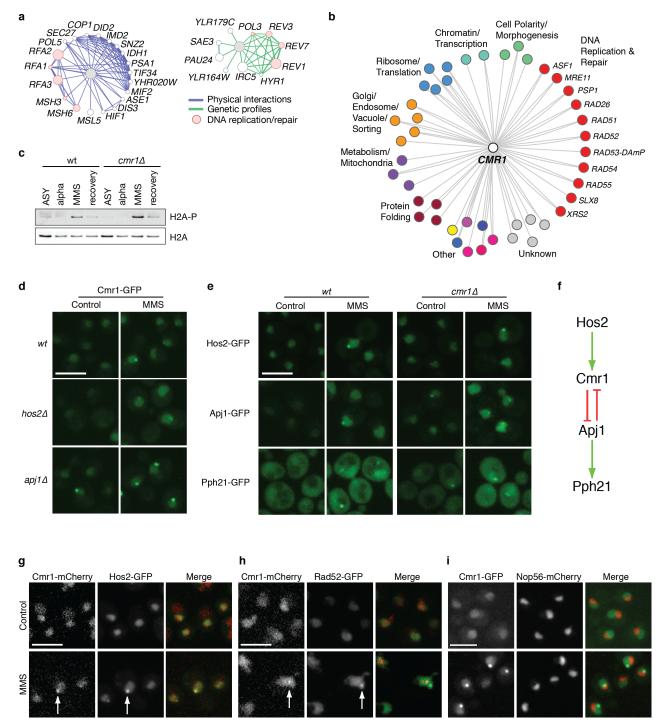
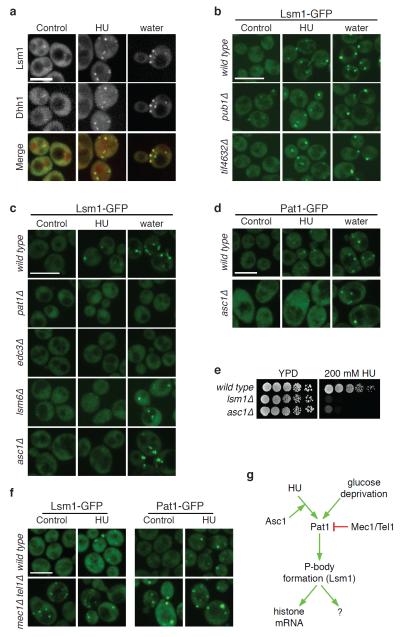
Comment in
-
Visualizing global effects of the DNA damage response.Nat Cell Biol. 2012 Sep;14(9):900-1. doi: 10.1038/ncb2571. Nat Cell Biol. 2012. PMID: 22945255
References
-
- Soufi B, et al. Global analysis of the yeast osmotic stress response by quantitative proteomics. Mol Biosyst. 2009;5:1337–1346. - PubMed
-
- Ptacek J, et al. Global analysis of protein phosphorylation in yeast. Nature. 2005;438:679–684. - PubMed
-
- Peng J, et al. A proteomics approach to understanding protein ubiquitination. Nat Biotechnol. 2003;21:921–926. - PubMed
References cited only in the Methods
-
- Brachmann CB, et al. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 1998;14:115–132. - PubMed
-
- Sherman F. Getting started with yeast. Methods Enzymol. 2002;350:3–41. - PubMed
-
- Winzeler EA, et al. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science. 1999;285:901–906. - PubMed
-
- Benschop JJ, et al. A consensus of core protein complex compositions for Saccharomyces cerevisiae. Mol Cell. 2010;38:916–928. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
