

Mitochondrial metabolism in Parkinson's disease impairs quality control autophagy by hampering microtubule-dependent traffic
- PMID: 22843496
- PMCID: PMC3471400
- DOI: 10.1093/hmg/dds309
Mitochondrial metabolism in Parkinson's disease impairs quality control autophagy by hampering microtubule-dependent traffic
Abstract
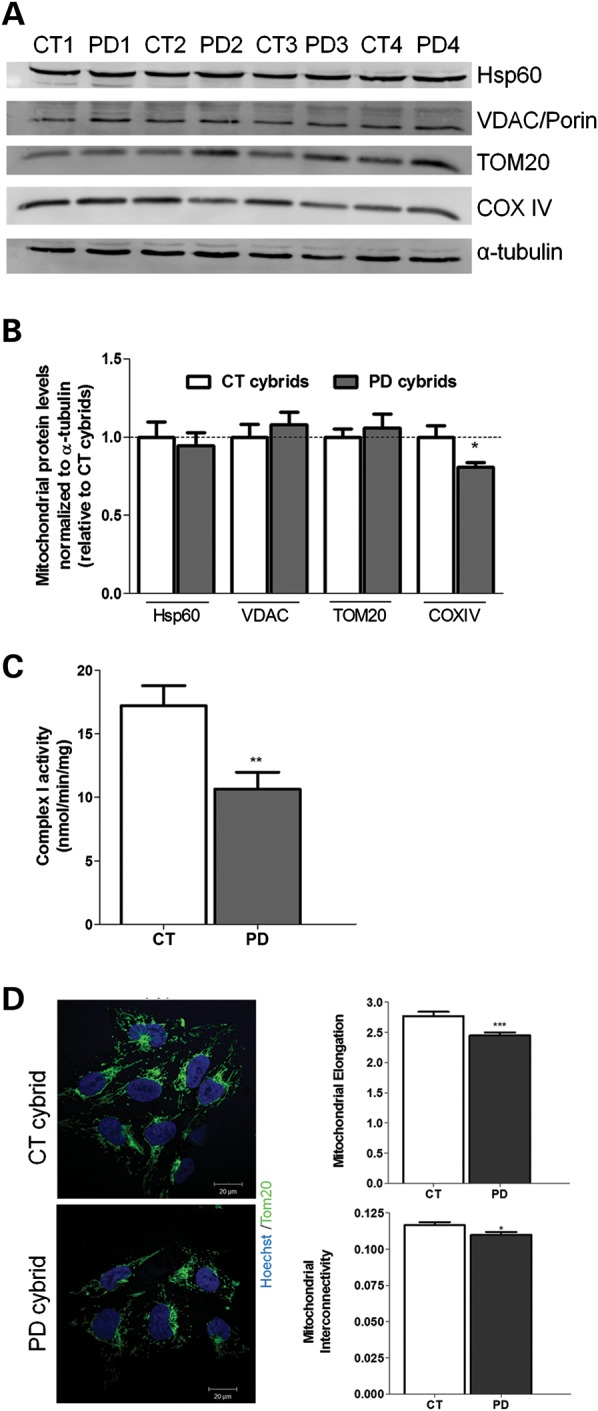
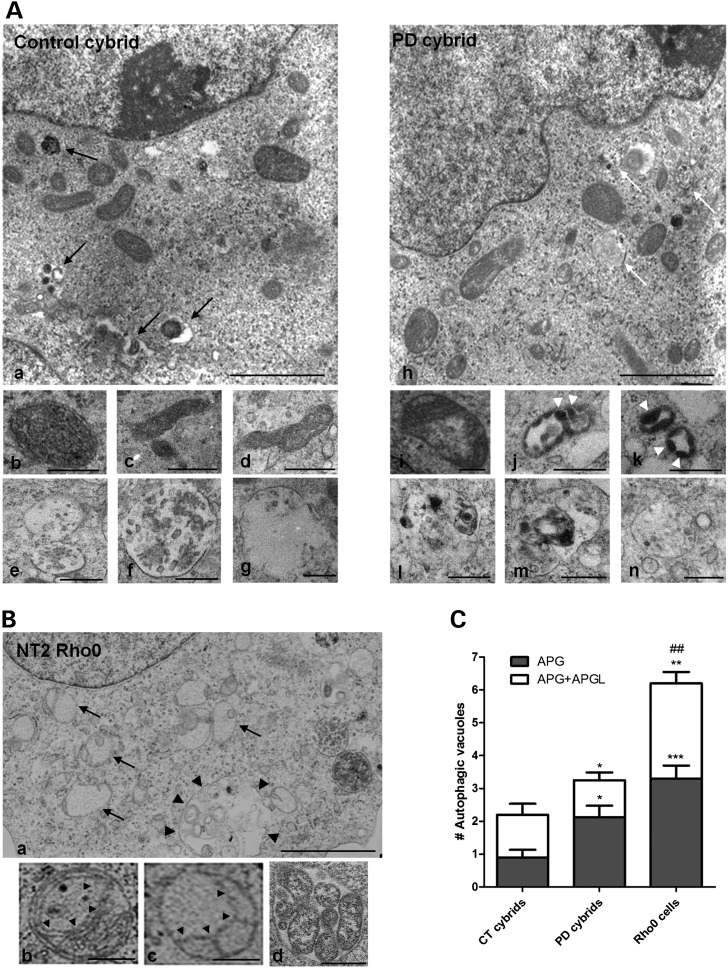
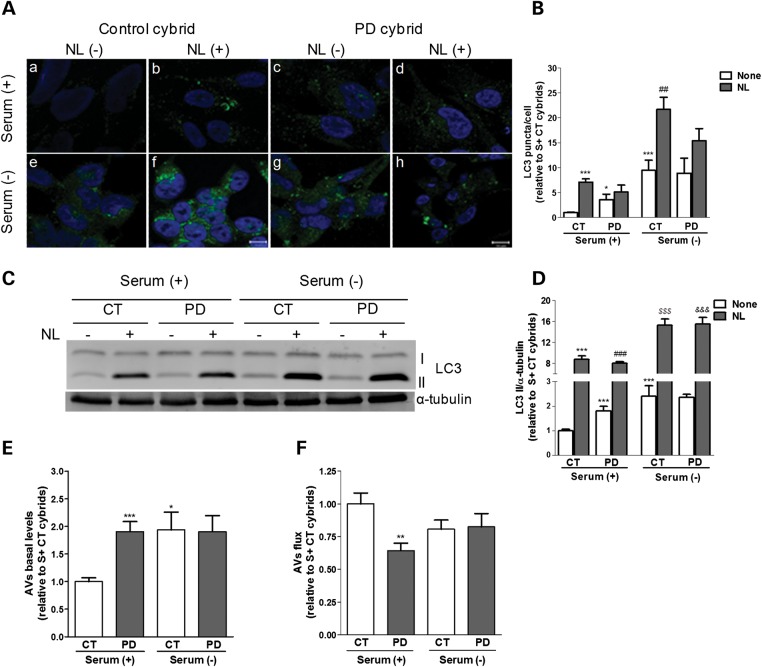
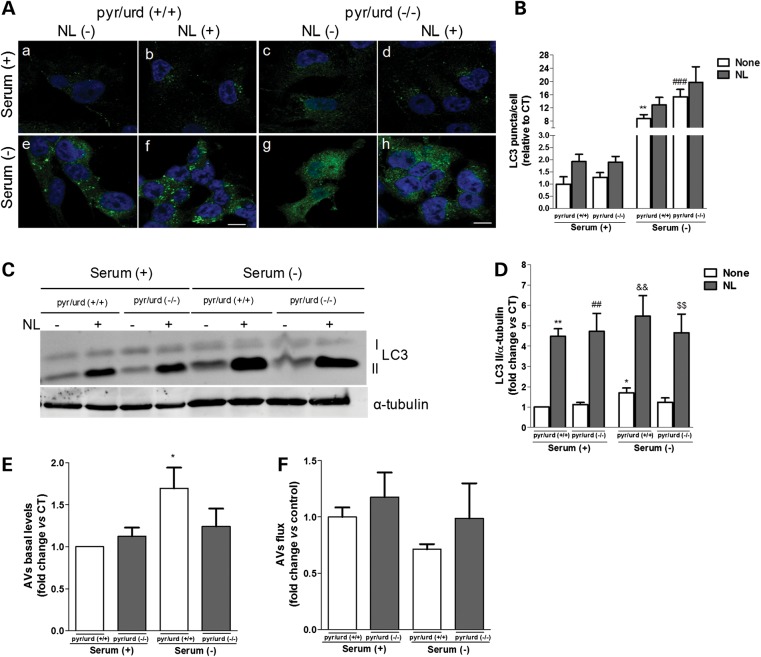
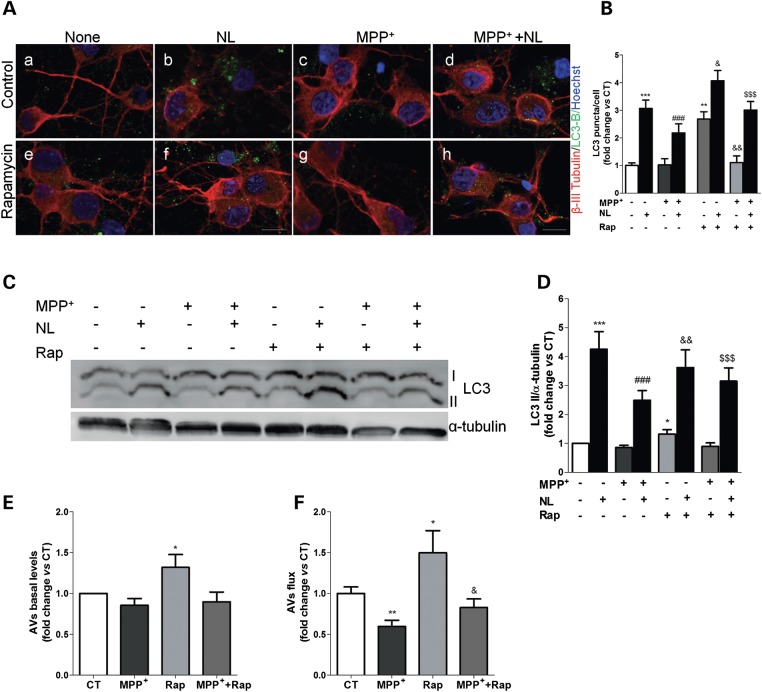
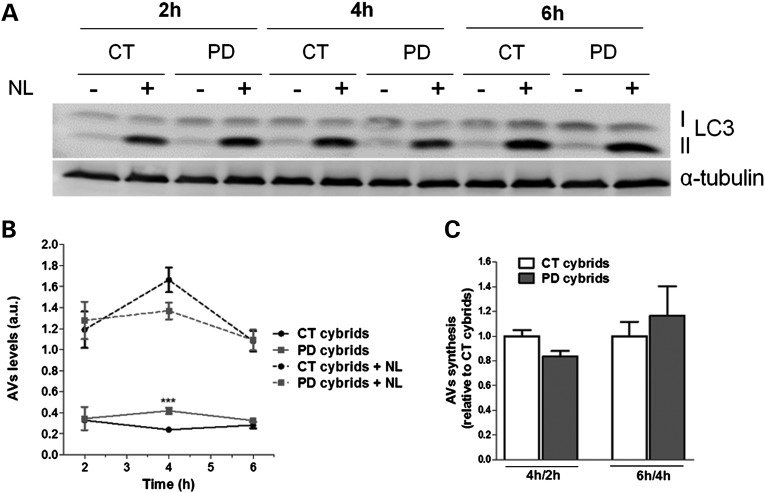
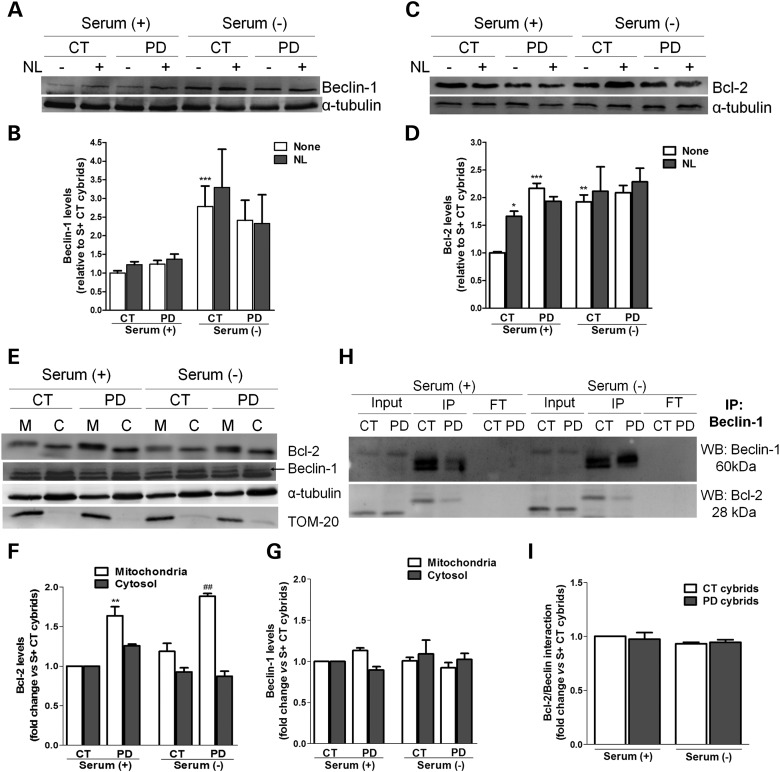
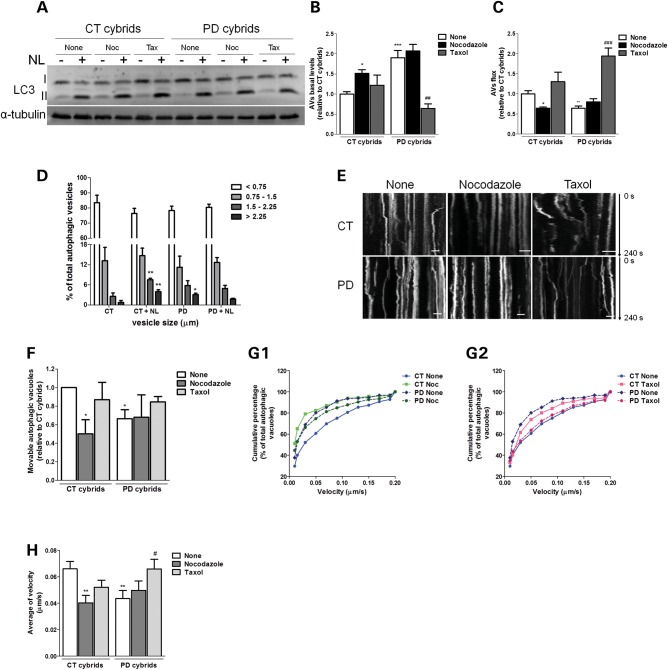
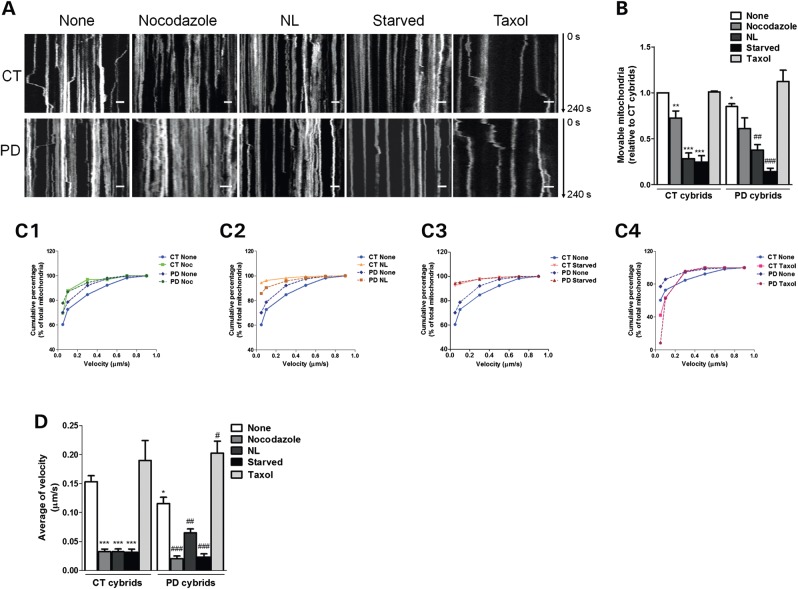
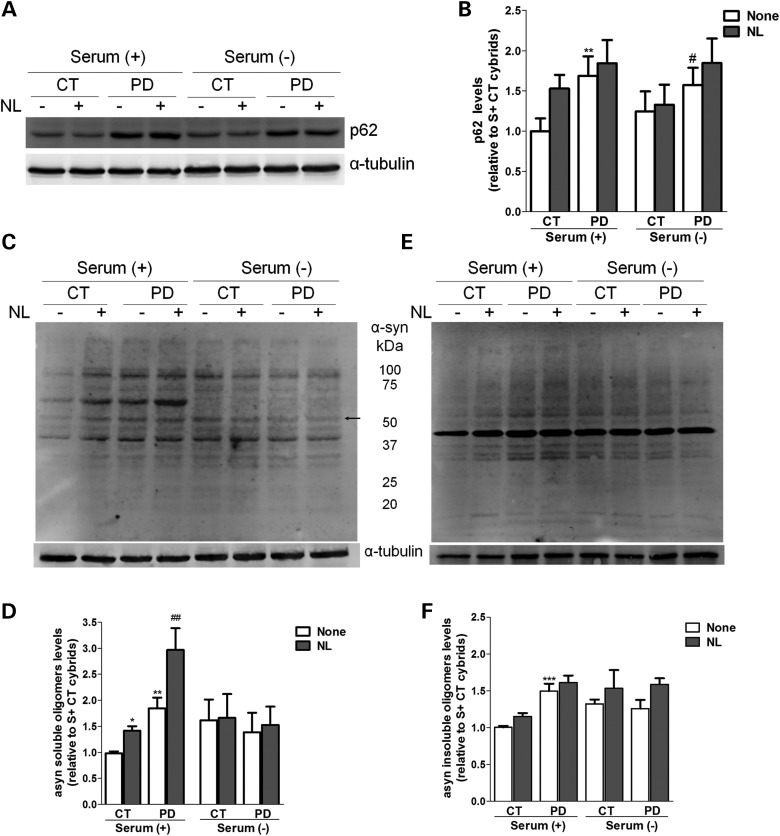
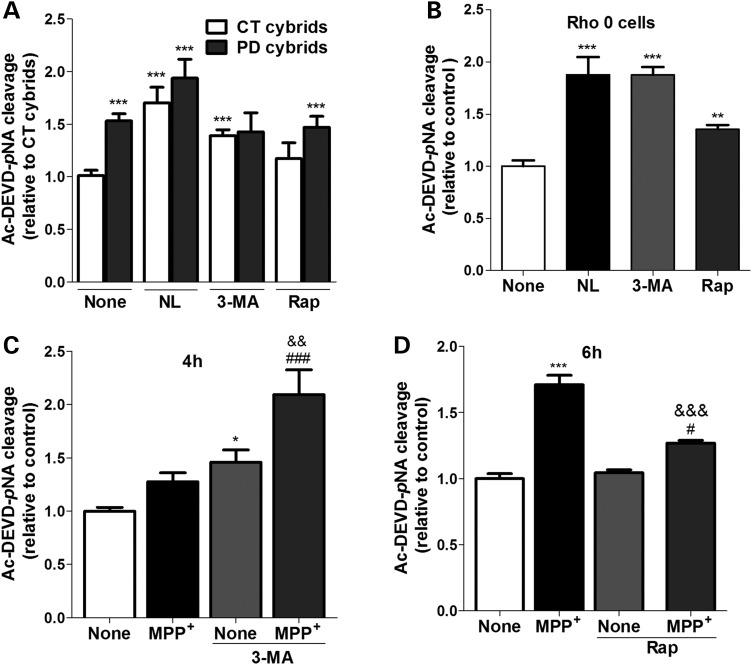
Abnormal presence of autophagic vacuoles is evident in brains of patients with Parkinson's disease (PD), in contrast to the rare detection of autophagosomes in a normal brain. However, the actual cause and pathological significance of these observations remain unknown. Here, we demonstrate a role for mitochondrial metabolism in the regulation of the autophagy-lysosomal pathway in ex vivo and in vitro models of PD. We show that transferring mitochondria from PD patients into cells previously depleted of mitochondrial DNA is sufficient to reproduce the alterations in the autophagic system observed in PD patient brains. Although the initial steps of this pathway are not compromised, there is an increased accumulation of autophagosomes associated with a defective autophagic activity. We prove that this functional decline was originated from a deficient mobilization of autophagosomes from their site of formation toward lysosomes due to disruption in microtubule-dependent trafficking. This contributed directly to a decreased proteolytic flux of α-synuclein and other autophagic substrates. Our results lend strong support for a direct impact of mitochondria in autophagy as defective autophagic clearance ability secondary to impaired microtubule trafficking is driven by dysfunctional mitochondria. We uncover mitochondria and mitochondria-dependent intracellular traffic as main players in the regulation of autophagy in PD.
Figures
References
-
- Forno L.S. Neuropathology of Parkinson's disease. J. Neuropathol. Exp. Neurol. 1996;55:259–272. - PubMed
-
- Schapira A.H., Cooper J.M., Dexter D., Jenner P., Clark J.B., Marsden C.D. Mitochondrial complex I deficiency in Parkinson's disease. Lancet. 1989;1:1269. - PubMed
-
- Parker W.D., Jr, Boyson S.J., Parks J.K. Abnormalities of the electron transport chain in idiopathic Parkinson's disease. Ann. Neurol. 1989;26:719–723. - PubMed
-
- Swerdlow R.H., Parks J.K., Miller S.W., Tuttle J.B., Trimmer P.A., Sheehan J.P., Bennett J.P., Jr, Davis R.E., Parker W.D., Jr Origin and functional consequences of the complex I defect in Parkinson's disease. Ann. Neurol. 1996;40:663–671. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
