

Understanding Food and Drug Administration regulatory requirements for an investigational device exemption for sponsor-investigators
- PMID: 22847340
- PMCID: PMC3448842
- DOI: 10.2310/JIM.0b013e318262df40
Understanding Food and Drug Administration regulatory requirements for an investigational device exemption for sponsor-investigators
Abstract
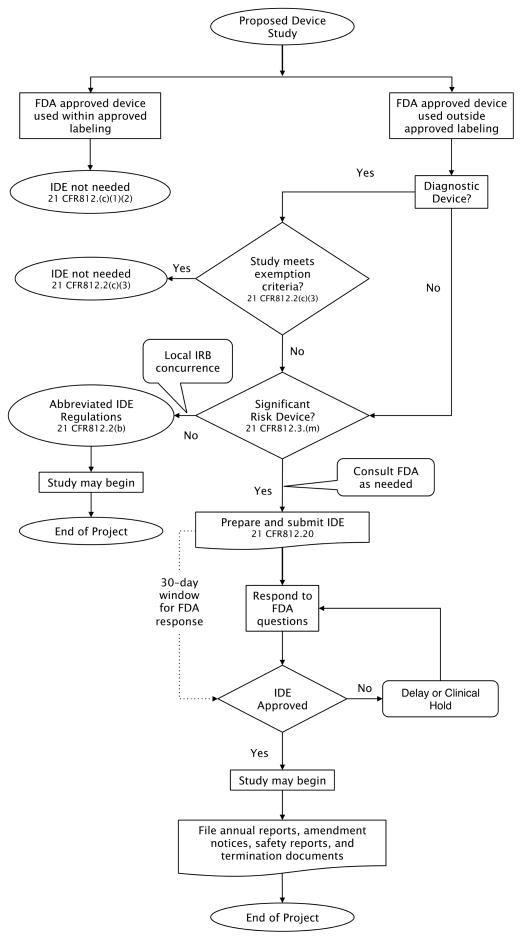
Clinical investigators in academic medical centers often perceive federal regulations as a significant obstacle to conducting clinical research. The regulatory authority of the Food and Drug Administration (FDA) extends to clinical studies of medical devices. Consequently, researchers wishing to conduct device research using FDA-approved as well as nonapproved devices must comply with federal regulations for investigational device exemptions (IDE) as described in Title 21 of the Code of Federal Regulations Part 812. FDA regulatory oversight is structured to match the risk to the subject to the risk of the device. Medical device studies can be categorized as follows: meeting exemption criteria, being a nonsignificant risk device, or being a significant risk device. All IDE studies must meet regulations for the protection of human subjects, but no additional federal filing on the part of the investigator is necessary for those that meet exempt criteria. Nonsignificant risk device studies require meeting abbreviated IDE regulatory requirements for the conduct of the study, but no previous FDA approval is required. Significant risk device studies require that the investigator also function as a sponsor and to file an IDE with the FDA for approval before starting. A sponsor-investigator filing an IDE follows the format and content described in 21 CFR 812.20. The study may begin 30 days after the date of submission receipt unless the FDA notifies the sponsor otherwise. While the IDE is active, the sponsor-investigator must meet the requirements for the conduct of the study and the required monitoring and reporting to the FDA.
Figures
References
-
-
Protection of Human Subjects. Title 45 C.F.R. Part 46. (2011)
-
-
-
Medical Devices. Title 21 C.F.R. Part 812 Subchapter H (2011)
-
-
-
Federal Food, Drug, and Cosmetic Act, 21 U.S.C. (2010 Edition).
-
-
-
Federal Food, Drug, and Cosmetic Act, 21 U.S.C. Subchapter II – Definitions (2010 Edition).
-
-
- [Accessed May 30, 2012.];Information Sheet Guidance For IRBs, Clinical Investigators, and Sponsors: Frequently Asked Questions About Medical Devices. 2006 Available at: http://www.fda.gov/downloads/RegulatoryInformation/Guidances/UCM127067.pdf.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
