

Scaling metagenome sequence assembly with probabilistic de Bruijn graphs
- PMID: 22847406
- PMCID: PMC3421212
- DOI: 10.1073/pnas.1121464109
Scaling metagenome sequence assembly with probabilistic de Bruijn graphs
Abstract
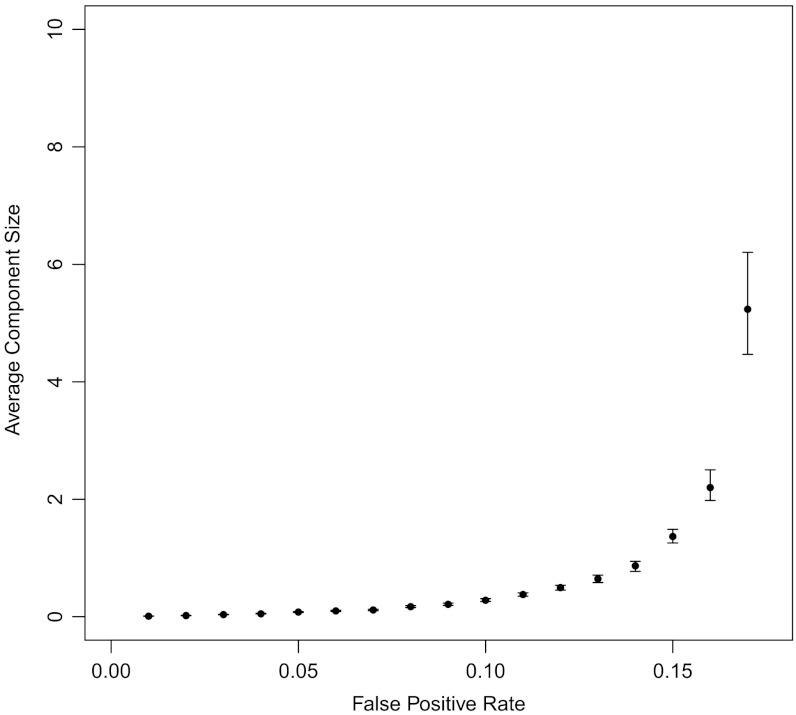
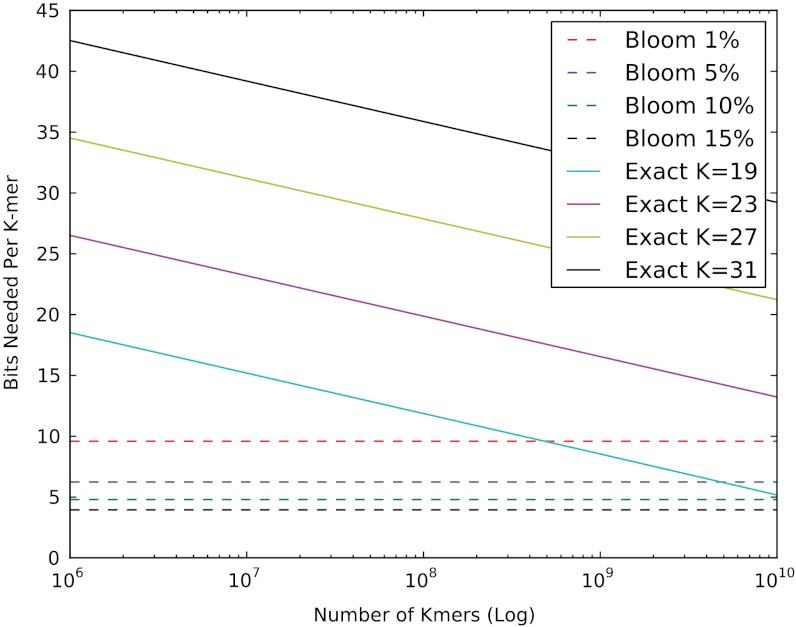
Deep sequencing has enabled the investigation of a wide range of environmental microbial ecosystems, but the high memory requirements for de novo assembly of short-read shotgun sequencing data from these complex populations are an increasingly large practical barrier. Here we introduce a memory-efficient graph representation with which we can analyze the k-mer connectivity of metagenomic samples. The graph representation is based on a probabilistic data structure, a Bloom filter, that allows us to efficiently store assembly graphs in as little as 4 bits per k-mer, albeit inexactly. We show that this data structure accurately represents DNA assembly graphs in low memory. We apply this data structure to the problem of partitioning assembly graphs into components as a prelude to assembly, and show that this reduces the overall memory requirements for de novo assembly of metagenomes. On one soil metagenome assembly, this approach achieves a nearly 40-fold decrease in the maximum memory requirements for assembly. This probabilistic graph representation is a significant theoretical advance in storing assembly graphs and also yields immediate leverage on metagenomic assembly.
Conflict of interest statement
The authors declare no conflict of interest.
Figures


References
-
- Hess M, et al. Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science. 2011;331:463–467. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
