

Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility
- PMID: 22848472
- PMCID: PMC3405143
- DOI: 10.1371/journal.pone.0041361
Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility
Abstract
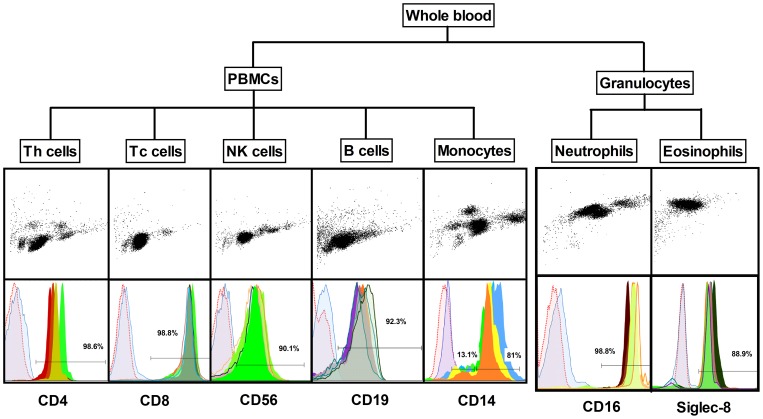
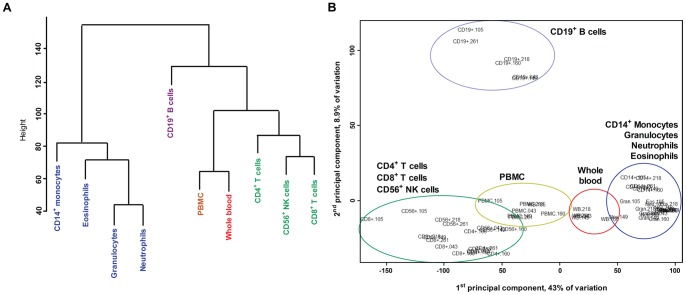
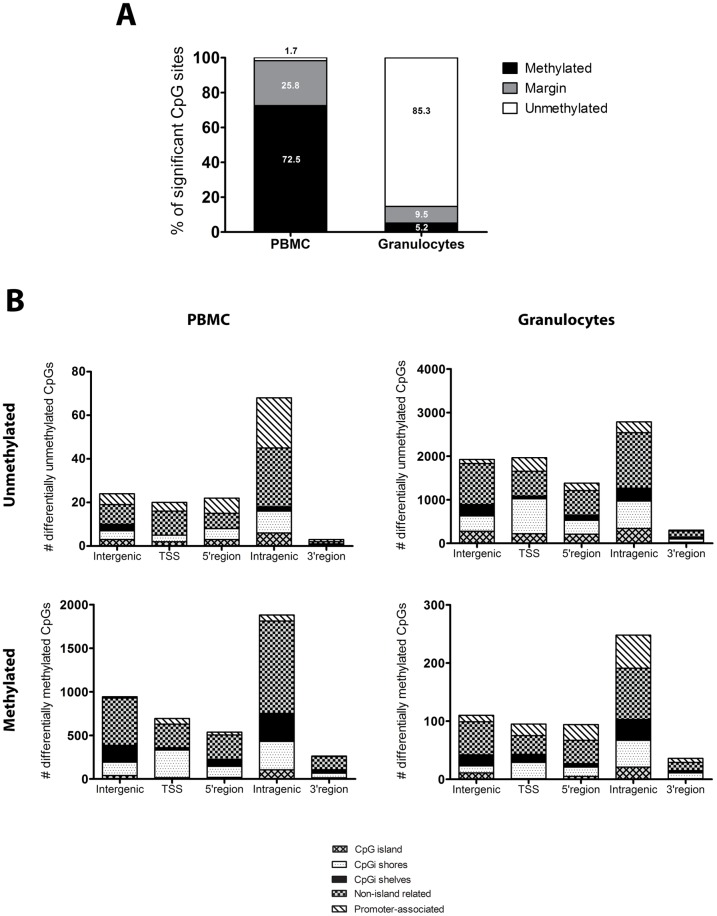
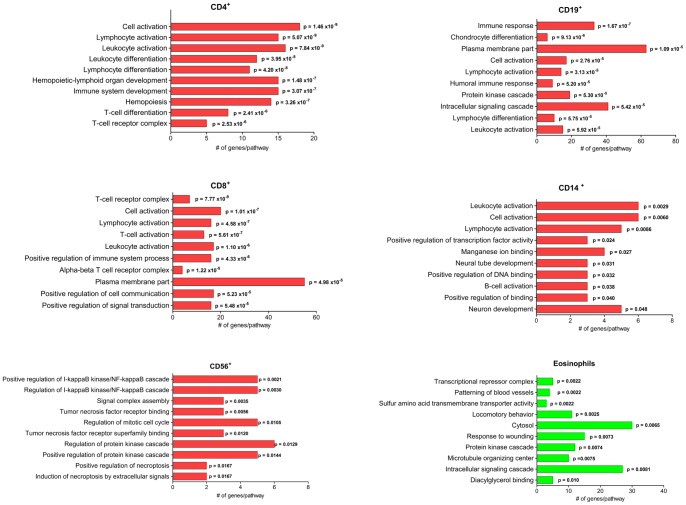
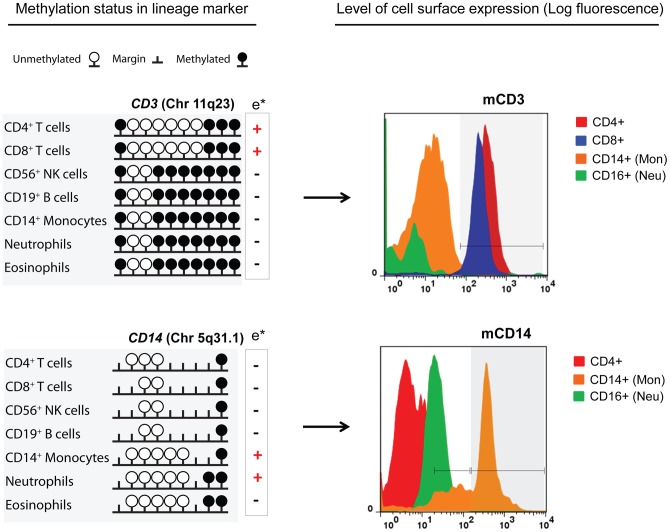
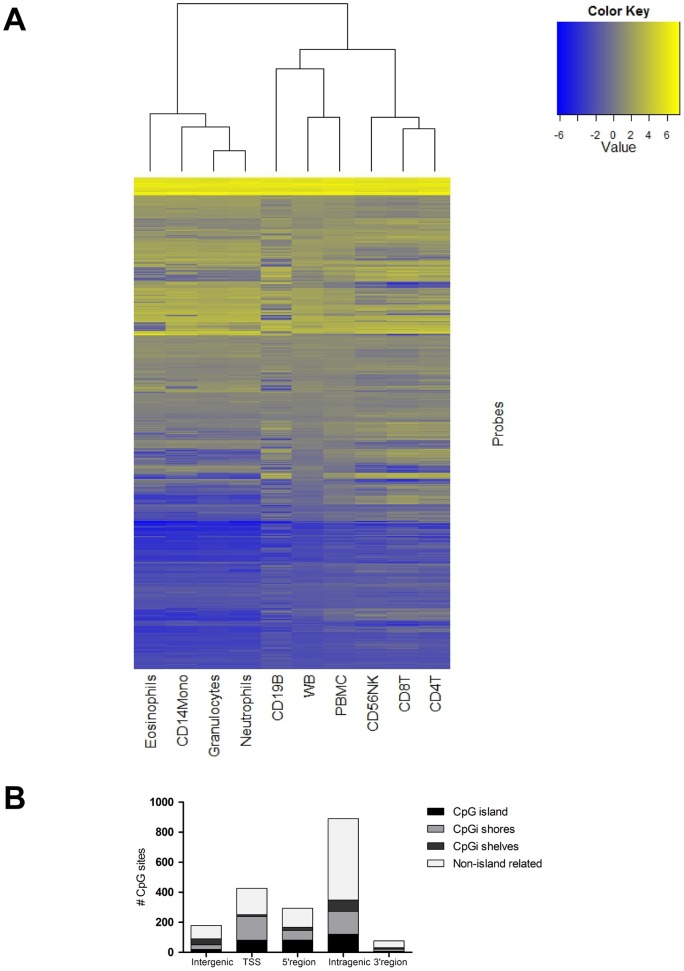
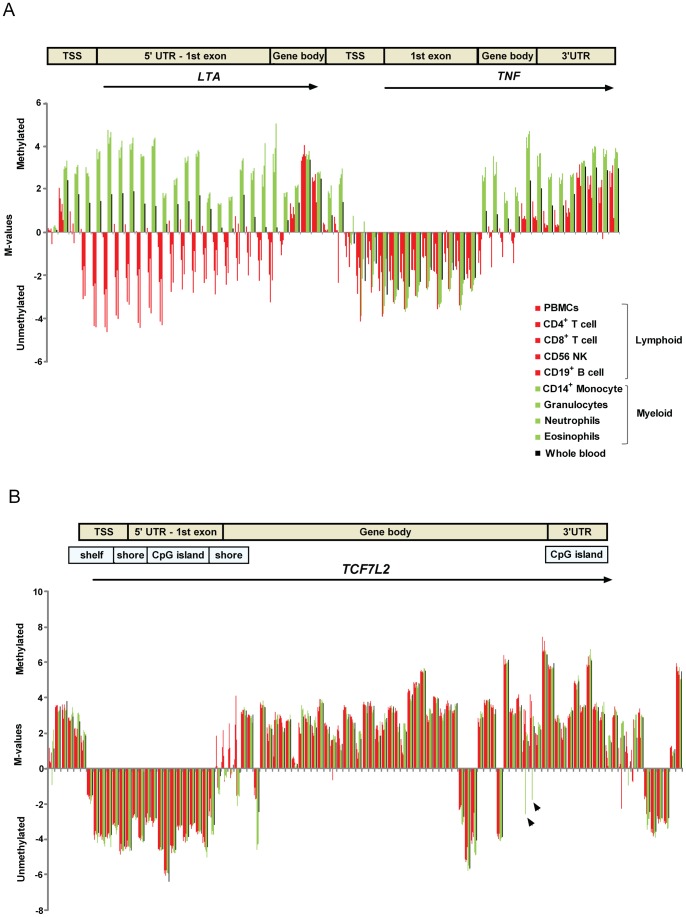
Methylation of cytosines at CpG sites is a common epigenetic DNA modification that can be measured by a large number of methods, now even in a genome-wide manner for hundreds of thousands of sites. The application of DNA methylation analysis is becoming widely popular in complex disorders, for example, to understand part of the "missing heritability". The DNA samples most readily available for methylation studies are derived from whole blood. However, blood consists of many functionally and developmentally distinct cell populations in varying proportions. We studied whether such variation might affect the interpretation of methylation studies based on whole blood DNA. We found in healthy male blood donors there is important variation in the methylation profiles of whole blood, mononuclear cells, granulocytes, and cells from seven selected purified lineages. CpG methylation between mononuclear cells and granulocytes differed for 22% of the 8252 probes covering the selected 343 genes implicated in immune-related disorders by genome-wide association studies, and at least one probe was differentially methylated for 85% of the genes, indicating that whole blood methylation results might be unintelligible. For individual genes, even if the overall methylation patterns might appear similar, a few CpG sites in the regulatory regions may have opposite methylation patterns (i.e., hypo/hyper) in the main blood cell types. We conclude that interpretation of whole blood methylation profiles should be performed with great caution and for any differences implicated in a disorder, the differences resulting from varying proportions of white blood cell types should be considered.
Conflict of interest statement
Figures
References
-
- Bocker MT, Hellwig I, Breiling A, Eckstein V, Ho AD, et al. Genome-wide promoter DNA methylation dynamics of human hematopoietic progenitor cells during differentiation and aging. Blood. 2011;117:e182–189. - PubMed
-
- Cedar H, Bergman Y. Epigenetics of haematopoietic cell development. Nat Rev Immunol. 2011;11:478–488. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
